What is Fabry Disease?:
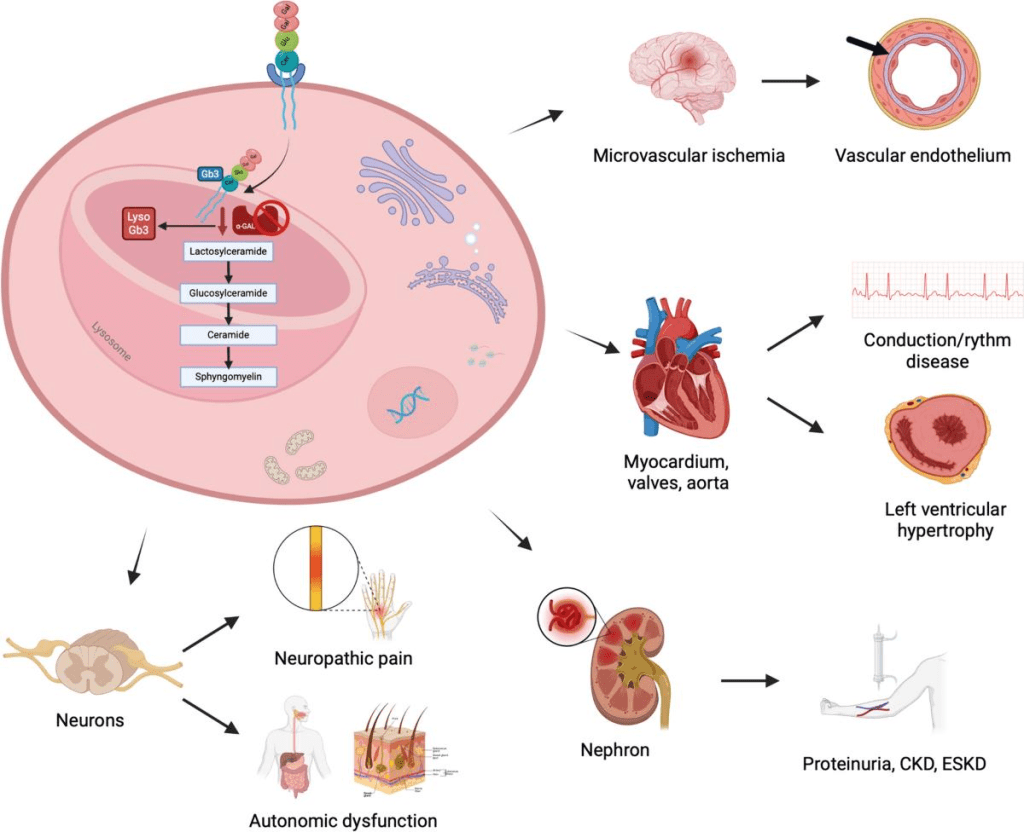
Fabry disease is a genetic, inheritable disorder affecting the metabolism of a certain fat. This is marked by a deficiency or significant lack of the lysosomal enzyme in the affected person’s body: α-galactosidase A (α-Gal A). This deficiency is caused by the mutations in the GLA gene, causing the body’s cells to improperly form α-Gal A. Lysosomes are the break-down centers of cells, responsible for digesting specific chemical compounds. α-Gal A is responsible for digesting glycolipids, specifically, globotriaosylceramide (GL-3). When the cells are deficient in α-Gal A, there is a resulting build up of GL-3 and other similar glycolipids in the cells. This tends to result in organ dysfunction and other abnormalities. This tends to affect the heart, kidneys, and blood vessels.
Since the GLA gene is bound to the X-chromosome, it is inherited as an X-linked, sex-bound disorder. Because of this, males are significantly worse affected by the disorder; however, females can still be affected, but they tend to be asymptomatic. It is still possible, though, for females to be equally as badly affected as males in their family.
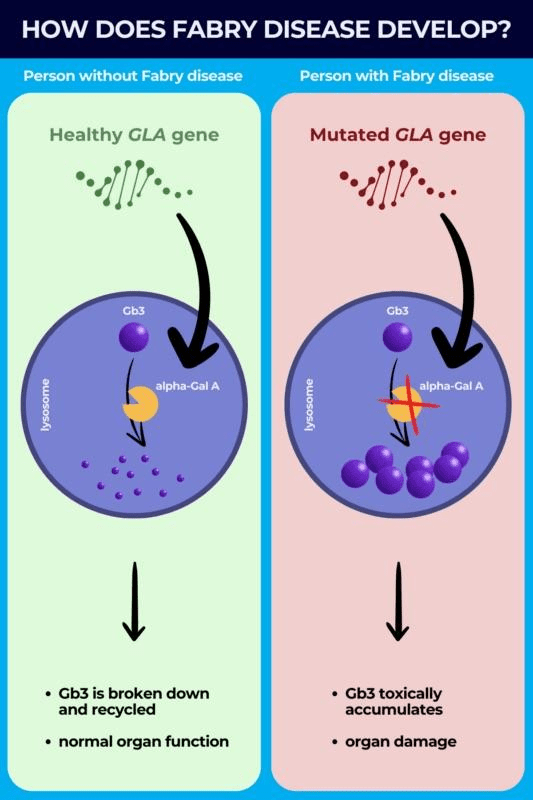
There are two different physical expressions of Fabry disease: type 1 and type 2, which is “later onset.” Both of these types tend to lead to kidney failure, cardiac failure, and/or premature death. Males with type 1 Fabry disease typically show symptoms starting in childhood and adolescence. This is marked by a severe accumulation of GL-3, which can cause worsening symptoms in old age. However, unfortunately, it is common for males to only live for 40 years with Fabry disease. Contrastingly, males who have “late onset” Fabry disease still retain some α-Gal A activity, and they lack the severity of GL-3 accumulation present in type 1 Fabry disease. Those with type 2 Fabry disease tend to have a normal childhood and adolescence, without symptoms. The normal symptoms of Fabry disease will usually present themselves between the ages of 20 and 69 years of age. Incidence of “late onset” Fabry disease varies based on ethnicity, race, and demography, but it is five to ten times more common than type 1 Fabry disease.
Symptoms:
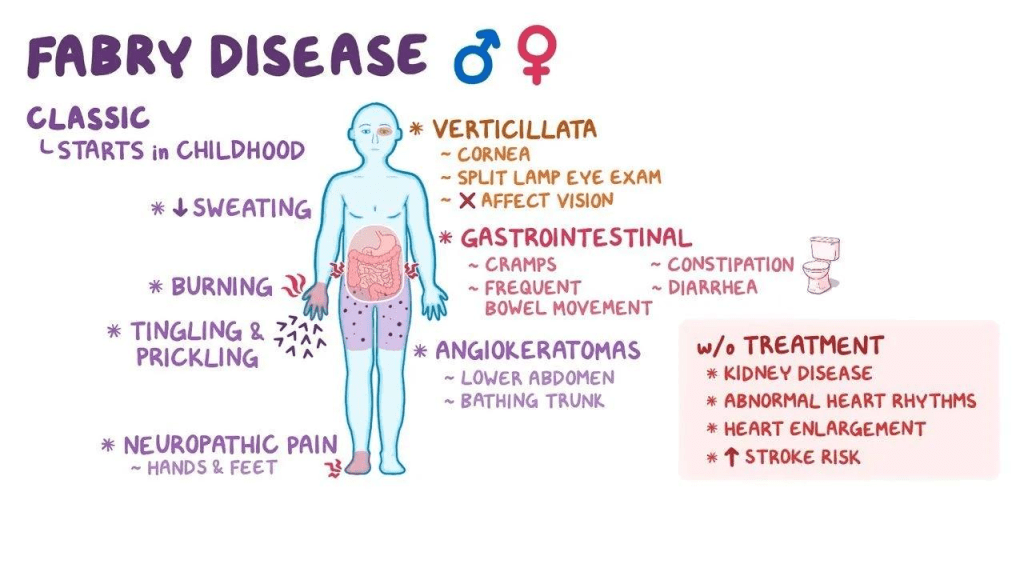
Type 1 Symptoms:
For type 1, some symptoms can present themselves as early as 2 years old. One of these symptoms is acroparethesia, which is the experience of a burning pain in the hands and/or feet that can also be associated with fevers. These acroparethesia episodes are often caused by exercise, stress, or fatigue. Another, slightly less severe symptom is anhidrosis, which results in decreased sweat production and discomfort as a result of warm temperatures. Another symptom only present in type 1 Fabry disease is the presence of a reddish/dark blue skin rash near the hips and knees, the rash may also be present near the umbilical area or the genitals in males with type 1 Fabry disease. Another common early symptom is gastrointestinal issues. This could include abdominal cramping, frequent bowel movements, and diarrhea could occur, most often after a large meal. Individuals with type 1 Fabry disease have abnormally large deposits of fats in their corneas, causing the foggy, whorl-like opacity of the eye; however, this does not affect their vision, and the individual should proceed to see normally. Some other symptoms of type 1 patients include fatigue, dizziness, headaches, weakness, nausea, and/or vomiting, delayed puberty, sparse hair growth, and on rare occasion, malformed joints of the fingers.
Type 1 and Type 2 Symptoms:
When individuals reach adulthood, the buildup of GL-3 leads to kidney and/or heart failure. The signs of development of kidney or heart disease could be renal dysfunction due to a build up of GL-3 in the smooth muscle cells of the kidney. In type 1 patients, their decline in kidney health begins young, and they typically need dialysis or a transplant by ages 35 to 45. In type 2 males, however, kidneys only become affected in the patient’s 30’s or later. Cardiac disease is also possible in both types of the disease, affecting the valves, nerves, arteries, heart enlargement, and more. Type 1 patients are diagnosed between their early 20s to 40s, while type 2 patients are typically diagnosed in their late 30s to 40s. Due to the build up of GL-3 in the heart and blood vessels in the brain, those with type 1 Fabry disease can experience hemorrhagic strokes later in life. Respiratory problems are also possible as a result of GL-3 build up in the lung tissue and cause serious bronchial lung disease. Other possible symptoms could be hearing loss, tinnitus, dizziness, and vertigo due to GL-3/Gb3 deposition, which are common in older, matured patients. Additionally, many males with type 1 Fabry disease were reported to have severe depression.
Causes:
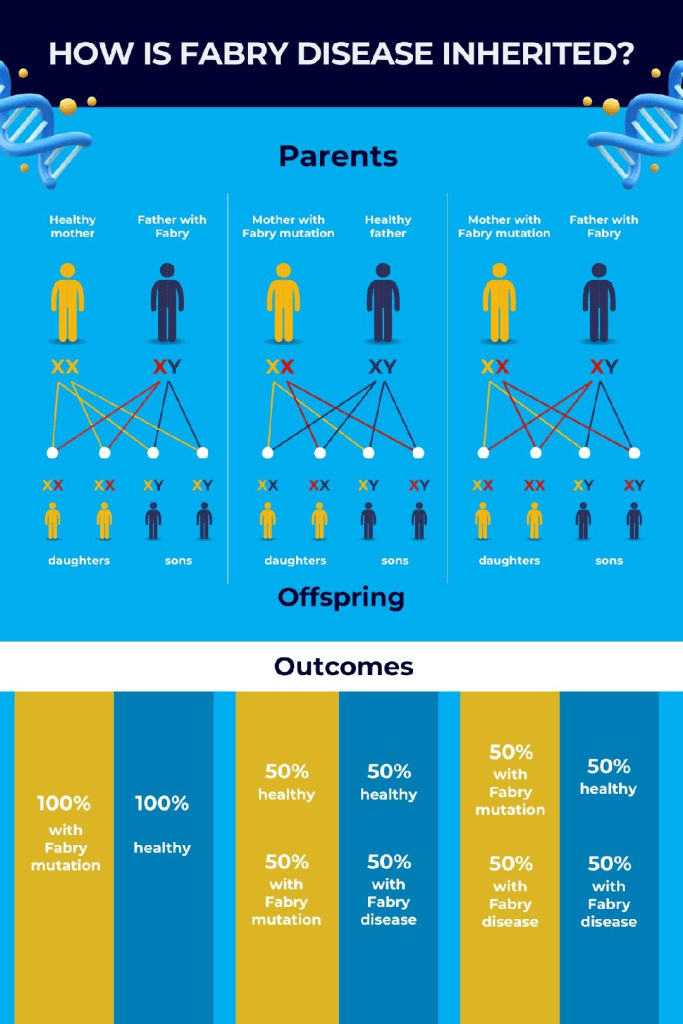
Fabry disease is a result of mutations to the GLA gene on the X-chromosome. As a result of the secondary X chromosome in females being “turned off,” the disease and its associated symptoms can be masked or reduced, resulting in most females with Fabry disease being asymptomatic. With Fabry disease, the severity of organ damage or how affected certain organs are depends on how many cells are controlled by the GLA gene. This helps to explain why Fabry disease in females is less consistent, since one X-chromosome is not active. Considering the fact that males only have one X-chromosome, if they have the GLA gene on their sole X-chromosome, they will have the disorder. This also goes for both type 1 and type 2 males, in contrast to their female counterparts whose symptoms range from asymptomatic to full expression based on how their X-chromosome activity.
Males with Fabry disease will always transmit the disease to their daughters and not their sons. Females, however, have a 50% risk of transferring the disease to their children with each pregnancy, this goes for any pregnancy, regardless of the gender of the child.
Currently, there are 965 known mutations of the GLA gene that cause Fabry disease, these mutations causing either of the two types of the disease. As a result, the severity and specific symptoms vary from family to family. Depending on how much the different mutations inhibit the activity of the α-Gal A enzyme, the type of Fabry disease will be different. It is also possible for mutations of the GLA gene to not cause Fabry disease.
Diagnosis:
The clinical diagnosis of Fabry disease can be made by physicians who recognize the characteristic symptoms, especially during an episode of pain in an individual. The most severe symptoms like renal failure and cerebrovascular disease only present themselves later on in adulthood. Unfortunately, because of this, type 2 males often miss their diagnosis, but it can still be made upon development of such diseases. This later diagnosis is typically done in a cardiac or stroke clinic. However, the diagnosis for both types is done by identifying the GLA gene mutation as well as the low α-Gal A enzyme concentration. Females tend to be diagnosed through noticing a mutation in their GLA gene.
Prenatal diagnosis is possible at around 10 to 15 weeks of pregnancy through α-Gal A enzyme and GLA gene mutation analyses through collections of villus samples. It is also possible to determine a family’s specific gene mutation, which allows for preimplantation genetic diagnoses. Newborn screening can identify affected males via the identification of lowered α-Gal A activity from the dried blood spots, followed by GLA gene sequencing.
Treatment:

Since Fabry disease causes multiple organ failure and dysfunction, most patients require extremely thorough treatment plans tailored to their individual symptoms and level of lipid build up. Enzyme replacement therapy is the most important treatment currently available for Fabry disease. With this therapy, enzymes are intravenously reintroduced to the body to help break down some of the build up. The two available enzymes are agalsidase alpha and agalsidase beta. ERT is a substitute for the missing enzyme that is characteristic of Fabry disease. ERT thus far has shown to prevent renal failure and/or decline, especially if ERT is started before any kidney damage has begun. ERT is the current recommendation for males with type 1 Fabry disease at any age. Certain oral therapies are being produced in the EU and the US to treat Fabry disease. It’s a drug that’s able to enhance the remaining enzyme activity for type 2 males. Certain clinical studies have proved the effectiveness of this study. Everything else should be treated symptomatically after consulting with the proper physician, but transplants may be required for the more severe cases of kidney failure and heart disease. It is suggested for families with known counts of Fabry disease to go through genetic testing.
How You Can Make a Difference:
The potential for innovative treatment is reliant on modern research which currently has limitations, rooted in the fact that there are a low number of cases. Clinical trials and further studies cannot occur without the proper funding. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. To learn more about Fabry disease, donation opportunities, or the progress being made on new treatments, visit the National Fabry Disease Foundation! The National Fabry Disease Foundation is an organization dedicated to supporting everyone with Fabry disease. They function to assist with Fabry disease education, facilitate Fabry disease diagnosis, provide assistance to individuals with Fabry disease, as well as support Fabry disease research and promote advocacy issues.
Let’s keep spreading awareness! – Lily
References:
Ganesh, J., & Desnick, R. J. (2019, June 6). Fabry Disease – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/fabry-disease/

Leave a comment