What is Sickle Cell Disease?:
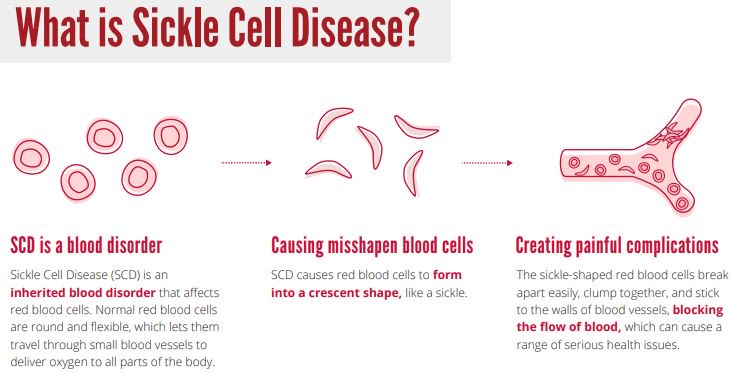
Sickle cell disease is an autosomal recessive disease affecting the blood. The characteristic trait is having sickle, crescent-shaped red blood cells circulating throughout the body’s bloodstream. These malformed blood cells very easily clump up with other blood cells, causing clots that block blood flow to the capillaries. This can prevent oxygen and nutrients from getting to all the places in the body they should go. The most common symptoms of SCD are excruciating chest and bone pain, infections, anemia, and potentially jaundice. The blood clots can cause organ disease and even failure, as well as stroke.
Symptoms:
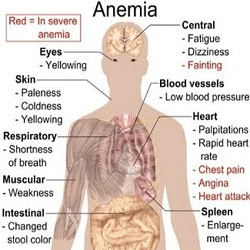
Many of the symptoms experienced by people with SCD are a result of the malformed hemoglobin in the red blood cells. The hemoglobin is a protein found in the blood, which is iron-rich and carries the blood from the lungs to the remainder of the body. When the hemoglobin is misshapen, it turns the normal red blood cells into a sickle shape, blocking the blood flow. The common symptoms of sickle cell disease are fatigue, anemia, and general pain. Episodes of SCD most commonly occur in the bones and the abdomen; however, they can happen about anywhere. The pain can be either acute or chronic as a result of clotting that can cause damage to the brain, lungs, kidneys, and joints. In infants the most obvious signs of SCD are swollen hands and feet, irritability, crying, and infections. Infections are mostly seen in infants, rather than adults.
The spleen may also become enlarged in cases of SCD as a result of blood cells getting trapped there. This damage to the spleen may cause inefficiency in fighting off seemingly mild infections, as it is unable to filter out blood cells or kill bacteria properly. Additionally diseases like acute chest syndrome, a life-threatening disease, can also arise as a result of SCD causing blocked blood flow to the lungs. This disease can result in fever, difficulty breathing, and intense chest pains. Other potential ailments resulting from SCD include strokes, even in young children, and painful, long-lasting erections for males.
The signs of sickle cell disease vary on a case by case basis, some patients hardly having any symptoms at all, while other individuals with more severe reactions may need hospitalization. Though SCD is present at birth, an individual may not express any symptoms for a few months to a few years after birth. SCD usually presents its symptoms within three years of age, typically as a result of a cold, overexertion, dehydration, and more. It is also not uncommon to see adolescents with SCD reaching puberty later and/or growing slower. As individuals with SCD age, more and more complications may arise, typically involving damaged blood vessels in the lungs, which, in turn, can cause shortness of breath and inability to exercise. Kidney damage, leg sores, vision problems, and joint damage are all especially common in older patients with SCD.
There are many forms of SCD, ranging from the severe sickle cell anemia to Sickle beta-plus thalassemia and sickle cell hemoglobin C disease, which are far less severe versions of the disease. Thus, it is very important for doctors to diagnose the exact form of SCD a person has; however, this can be very difficult as the differences between the versions are not always clear.
Causes:

The most direct cause of SCD is the mutation of both copies of the gene responsible for encoding the production of beta-globin: hemoglobin beta (HBB) gene. Beta-globin is a component of the larger protein, hemoglobin, which consists of two units of beta-globin and two units of a different protein, called alpha-globin, formed by a gene called HBA. These units of alpha-globin and beta-globin all attach to an iron-based molecule called heme, but the heme is still able to bind to an oxygen molecule to transport it. This hemoglobin in the blood allows it to bind to the oxygen molecules in the lungs from the inhaled air and transport the oxygen to other parts of the body. In normal red blood cells contain hemoglobin A are soft, round, and last around 120 days.
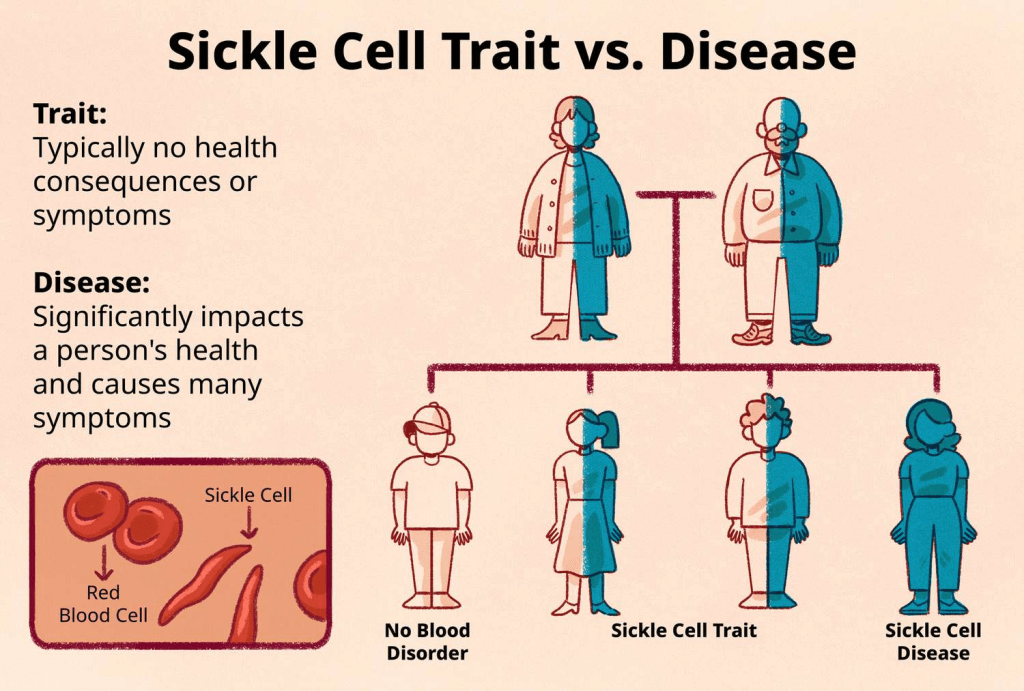
Sickle cell anemia (HbSS disease), which is the most common form of SCD, is caused by a very specific mutation in the HBB gene, resulting in the gene encoding the creation of malformed units of beta-globin called hemoglobin S (HbS) in the red blood cells. Individuals with sickle cell anemia inherited two disease-causing alleles from each parent, causing hemoglobin S to replace both of the normal beta-globin units present in healthy hemoglobin. The particular gene change replaces one amino acid in the sequence of the beta-globin protein, resulting in the abnormal sequence of hemoglobin S sticking together and forming long, rigid chains that cause the blood cells to bend in on themselves, which gives the blood cells the characteristic sickle-shaped appearance.
Unfortunately, sickled blood cells die much faster than regular red blood cells, only lasting about 16 days, causing serious anemia. Many parts of the body will be unable to receive the amount of blood they need, which is the main root of the worst complications of sickle cell anemia. Any different alteration to the HBB gene will cause the other kinds of SCD caused by a individual inheriting one copy of an altered hemoglobin gene from one parent and a copy of the disease-causing gene from their other parent. In these specific cases only one beta-globin unit is replaced with hemoglobin S, while the other beta-globin is replaced by a slightly different gene variant denoted by a specific letter like beta thalassemia, hemoglobin C, D, or E.
If an individual inherits only one normal HBB gene and one altered HBB gene (HbS), they are said to be carriers for the disease. One of their genes will express the normal hemoglobin, while the other gene will express the altered hemoglobin that causes the blood cells to clot and become sickle-shaped. Since these specific alleles have a codominant relationship, they will both be expressed throughout the body, meaning that the person will have a combination of normal and sickled blood cells. However, since there are still some normal blood cells produced, it is not enough for them to fully experience the effects of the disease. Though, in scenarios of high exertion, pressure, dehydration, or low oxygen, this person may experience SCD-affiliated pain.
However, it is important to note that the sickle shape of affected blood cells protect against malaria. For example, if a person with SCD were to be bitten by a mosquito carrying malaria, they would be far less likely to contract the disease. This is a result of the shape of the affected blood cells, as the shape makes it much harder for the malaria to grow, protecting the individual. In some regions natural selection promotes the expression of SCD to protect against malaria, skewing the numbers of cases in certain demographics and climate zones.
Diagnosis:
Standard American protocol requires that all babies born in the United States must be screened for SCD within 24-48 hours after birth, in addition to other screenings. This specific test is a high-pressure liquid form of chromatography testing. If a baby comes back positive from this test it doesn’t necessarily mean they have the disease, but it means they are likely to and further genetic testing and verification must be done before a diagnosis can be made. These screenings are also crucial to making sure that as many complications as possible are limited from developing via new treatments.
Treatment:
The treatment for SCD focuses on the prevention of the complications associated with SCD as well as any episodes of pain that individuals may experience. These treatment methods include lifestyle changes, medical screenings, patient education, avoiding triggers, and more. It is extremely rare that a person can be cured of SCD, only very early screening and prevention techniques have the ability to drastically improve the quality of life for a person with SCD. Regularly scheduled checkups and educational meetings are crucial, as they can help a patient’s family more prepared, immensely improving the health of the individual. These educational sessions are especially important when the patient is a child, so his/her parents can monitor them for fevers. Low doses of penicillin combined with informing parents help to ensure patients get the care they need, decreasing instances of infection and death. There are also other simple changes one can make to their day to day life to help mitigate the effects of sickle cell disease, including hydrating regularly, staying in moderate temperatures, exercising, taking deep breaths, not getting fatigued, as well as avoiding any physical trauma.
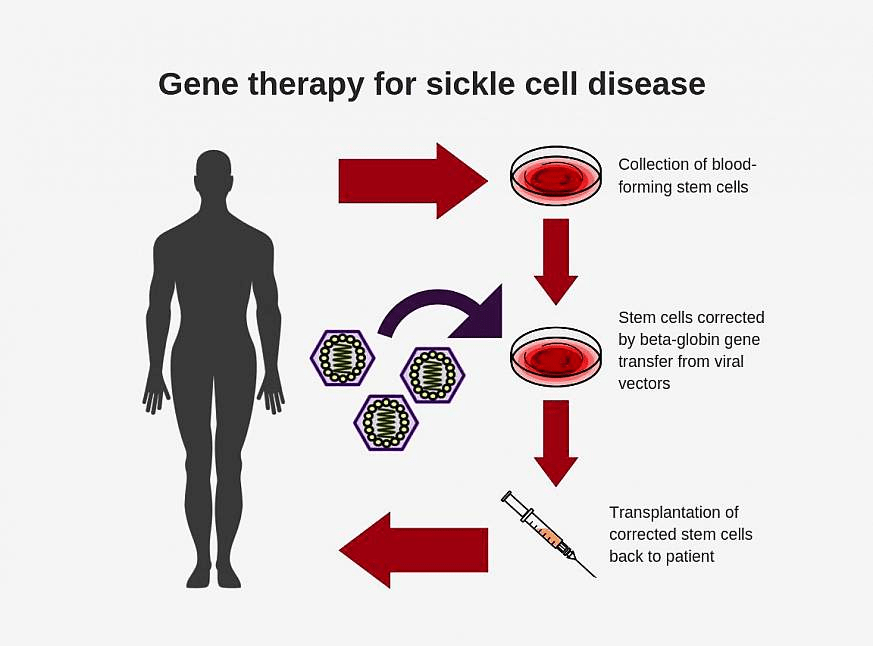
One thing that is most important to avoid is sickle cell pain and to treat it right when it does occur. Response methods to SCD pain include hydration, staying warm, walking, and breathing deeply. However, since the pain caused by SCD is very intense distraction and stimulation can be very helpful for individuals with SCD. These distractions include massages, acupuncture, and hypnosis. However, there are also some pain-reliever medicines that can be of a lot of help to patients such as non-steroidal anti-inflammatory agents and opiate analgesics. Blood transfusion is also used in the most dire of circumstances, specifically in cases of severe anemia, before surgery, and to reduce the risk of or treat a stroke. Some individuals may have to undergo surgery to organs like the gall bladder to remove gallstones as a result of damage from lack of proper blood flow. Stem cell transplants have been suspected as a cure for SCD; however, the success rate of this operation varies greatly based on several risk factors, making the likelihood for success rather low.
There are many drugs currently being tested by the FDA. For example folic acid has been used to make sure that the body has enough red blood cells to function properly. However, most of these drugs may only be administered to a child with SCD after he/she reaches a certain age, typically around age 12. Additionally, genetic counseling and therapies may be recommended for a family to undergo after a case of SCD was reported in the family.
How You Can Make a Difference:
Though sickle cell disease is prevalent in many African and South Asian countries, there is still no foolproof cure or treatment available for these people. Without the appropriate funding to further research and clinical trials on a treatment for sickle cell disease, many people will go without the care they need. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about SCD, donation opportunities, or the progress being made on new treatments, visit the Sickle Cell Disease Foundation! The Sickle Cell Disease Foundation aims to educate, screen, and counsel those at risk of having children with sickle cell disease. The SCDF focuses on helping those with SCD get the care they deserve.
Let’s keep spreading awareness! – Lily
References:
Alyea, G., & Bender, M. (2024, March 14). Sickle Cell Disease – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/sickle-cell-disease/

Leave a comment