What’s Cri du Chat Syndrome?:

CdCS is a genetic disorder caused by the random deletion of a section of the fifth chromosome. Since the deletion is random, symptoms vary in each person based on where the section was deleted from and how much was deleted. However, the most common symptoms include cat-like cries, distinct facial features, delayed growth, and an abnormally small head. Affected children tend to have moderate to severe intellectual disabilities, in addition to delays in skills development. The random error causing CdCS occurs early on during development inside the womb. Current treatment is symptomatic and may include services such as therapies and surgeries.
Symptoms:
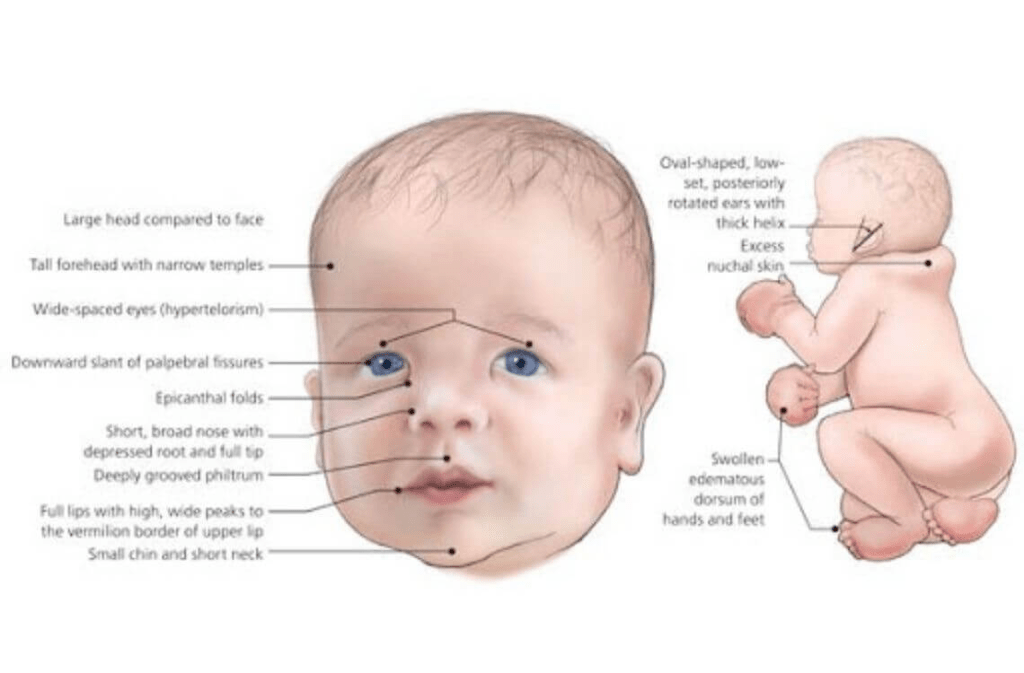
The most characteristic symptom of CdCS is a high-pitched cry, similar to the mew of a cat, especially during the first few weeks of life. However, this cry becomes less and less noticeable as the child ages. The symptoms are different depending on how much of the chromosome is deleted, as well as where the deletion occurred. These symptoms include low birth weight, growth deficiencies, low muscle tone, as well as an abnormally small head. Some distinct facial features of children with CdCS include having a remarkably round face, wide space between eyes, crossed eyes, downward slanting eyelid folds, downturned corners of the mouth, low-set ears, abnormally small-sized jaw, little space between the upper lip to the nose, opening or split in the roof of the mouth, opening or split of the upper lip, and/or a full lower lip.
Additional symptoms include a middle cleft in the uvula, scoliosis, recurrent ear infections, resulting in a high risk of hearing loss, heart defects, inguinal hernia, gastroesophageal reflex, abnormalities in the kidney and urinary tract, respiratory difficulties, webbed fingers and toes, bent or curved of the pinkies, clubfeet, abnormal larynx, nearsightedness, cataracts, prematurely gray hair, as well as repeated respiratory and intestinal infections. Some genital abnormalities may include failure of the testes to descend, as well as an opening on the underside of the penis. As children age, their round faces may develop into long, narrow faces. In addition, what once was low muscle tone at birth, may develop into high muscle tone later on in life.
Some developmental issues that may develop are motor skill delays, such as delays in head control, sitting up, and walking, intellectual disability, delay in speech development, ability to understand speech better than they can communicate it, hyperactivity, self-injurious behavior, repetitive movements, gentle personality, obsessive attachment, feeding and eating difficulties.
Causes:
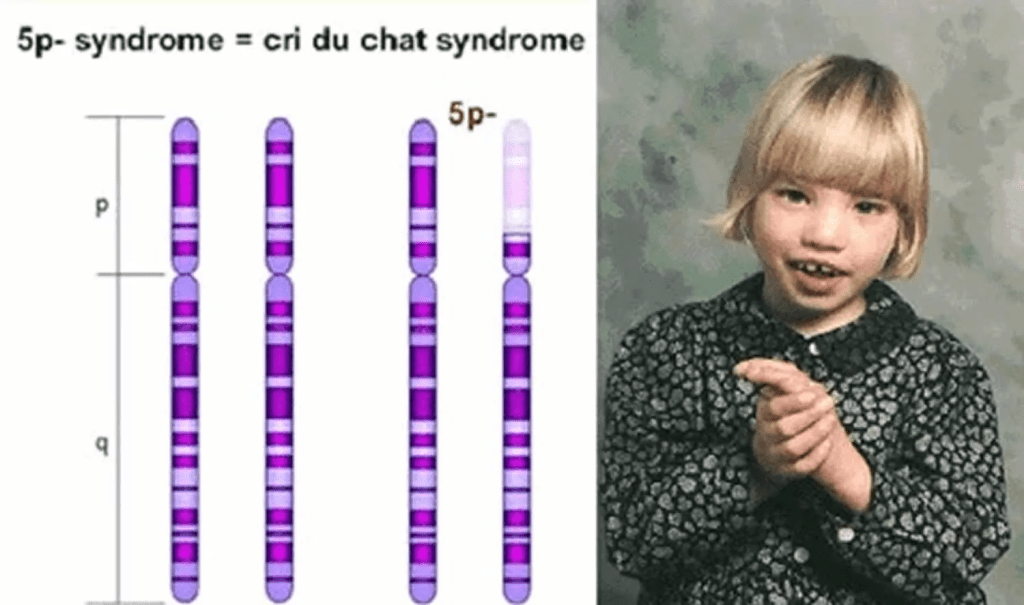
CdCS is a chromosomal disorder resulting from the partial deletion of part of the short arm (p) of the fifth chromosome. Chromosomes are divided into two arms based on their location relative to the centromere. As a result the shorter arm is known as “p”, and the larger arm is “q”. Because of this, CdCS is also called 5p-, as there is missing genetic material from the p arm of the fifth chromosome.
Researchers determined that some symptoms are associated with certain regions on the p arm of chromosome 5. Because of this, they have been able to identify genes that could be responsible for the development of CdCS. Examples include the TERT gene and the SEMA3F gene. If these genes are deleted, a wide range of characteristic CdCS-related features develop. Another CdCS-causing gene is the CTNND2 gene. By deleting this gene, more intellectual disability will arise, as it is linked to other genes. If researchers can determine which specific phenotypes are linked to which to specific deletions in the fifth chromosome, it may aid in the diagnosis and management of the disease.
Most cases of CdCS seem to occur randomly very early in embryonic development. Most deletions are paternal in origin, resulting from improper sperm formation in the child’s father. Parents of a child with this random deletion tend to have normal chromosomes with a low chance of having another child with CdCS.
Approximately 10-15% of patients have CdCS caused by translocation involving chromosome 5p and a different chromosome. In a balanced translocation, pieces of chromosomes from two different chromosomes swap places with each other, but no material is lost or gained. Thus, when a person has a balanced translocation they do not have any problems since they do not have an excess nor lack of genetic material. However, if a child inherits an affected chromosome 5, involved in a balanced translocation, the child now has a chromosome 5 that is missing genetic material, leading to developing CdCS. In addition, because, in this scenario, a parent can pass on an affected chromosome 5, there is a higher chance of having another child with CdCS. However, further chromosomal analysis can help determine whether a parent has had a balanced translocation or not.
Diagnosis:
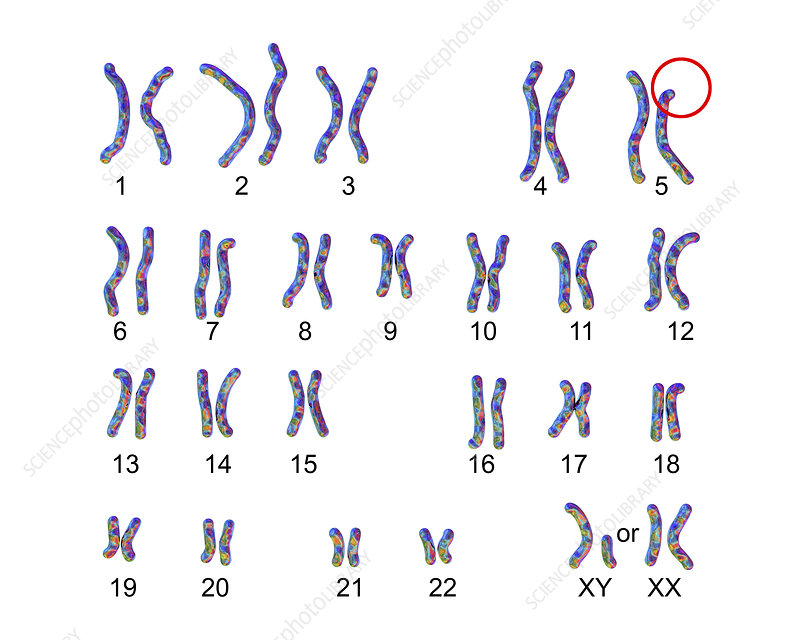
CdCS can be diagnosed both before and after birth. To be diagnosed prenatally, a procedure called amniocentesis must be conducted. Amniocentesis includes taking a sample of the amniotic fluid for testing. This fluid contains some cells from the fetus, holding the fetus’s chromosomes, which can be tested for any deletion in the fifth chromosome. This testing is paired with ultrasounds, which may reveal the physical features of CdCS.
In newborns, the diagnosis of CdCS can be made as a result of thorough clinical evaluation, identification of characteristic signs and identification of characteristic symptoms. The diagnosis can then be confirmed by karyotyping, revealing the deletion of the p arm of chromosome 5. Other tests are fluorescence in situ hybridization, comparative genomic hybridization, or a quantitative polymerase chain reaction. Chromosomal studies can also be performed on the parents, determining if one of them has had a balanced translocation.
Treatment:
CdCS treatment is completely symptomatic, requiring the efforts of a team of specialists. This team may include pediatricians, orthopedists, surgeons, cardiologists, speech pathologists, neurologists, dentists, physical and occupational therapists as well as other health care professionals to systematically and comprehensively plan a child’s treatment. Since some children with CdCS have sensory-neural deafness, auditory testing should be performed when appropriate.
Early intervention is extremely important to make sure children with CdCS reach their highest potential. Beneficial services include special remedial education, physical therapy, speech therapy, special services and more services. Most children with CdCS are enrolled in therapies, especially before they reach one year of age. Surgery may need to be performed to treat certain symptoms associated with CdCS, including congenital heart defects, strabismus, scoliosis, clubfoot, cleft palate and cleft lip.
The survival for children with CdCS is generally good, considering most CdCS deaths occur within the first year of life. Many affected individuals have lived beyond 50 years of age. Additionally, genetic counseling is recommended for affected individuals and their families.
How You Can Make a Difference:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures or legitimate medicines for the disease, many more children will go on to develop Cri du Chat syndrome. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about CdCS, donation opportunities, or the progress being made on potential treatments, visit the Cri Du Chat Research Foundation! The Cri Du Chat Research Foundation strives to “accelerate translational research focused on using innovative technologies to develop therapeutics to cure the major neurologic symptoms associated with 5p- syndrome in order to improve the lives and independence level of every child and adult in the world living with this rare disease.”
Let’s keep raising awareness! – Lily
References:
Chauhan, A., & Alyea, G. (2024, July 23). Cri du Chat Syndrome – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/cri-du-chat-syndrome/

Leave a comment