What is Cystic Fibrosis?:
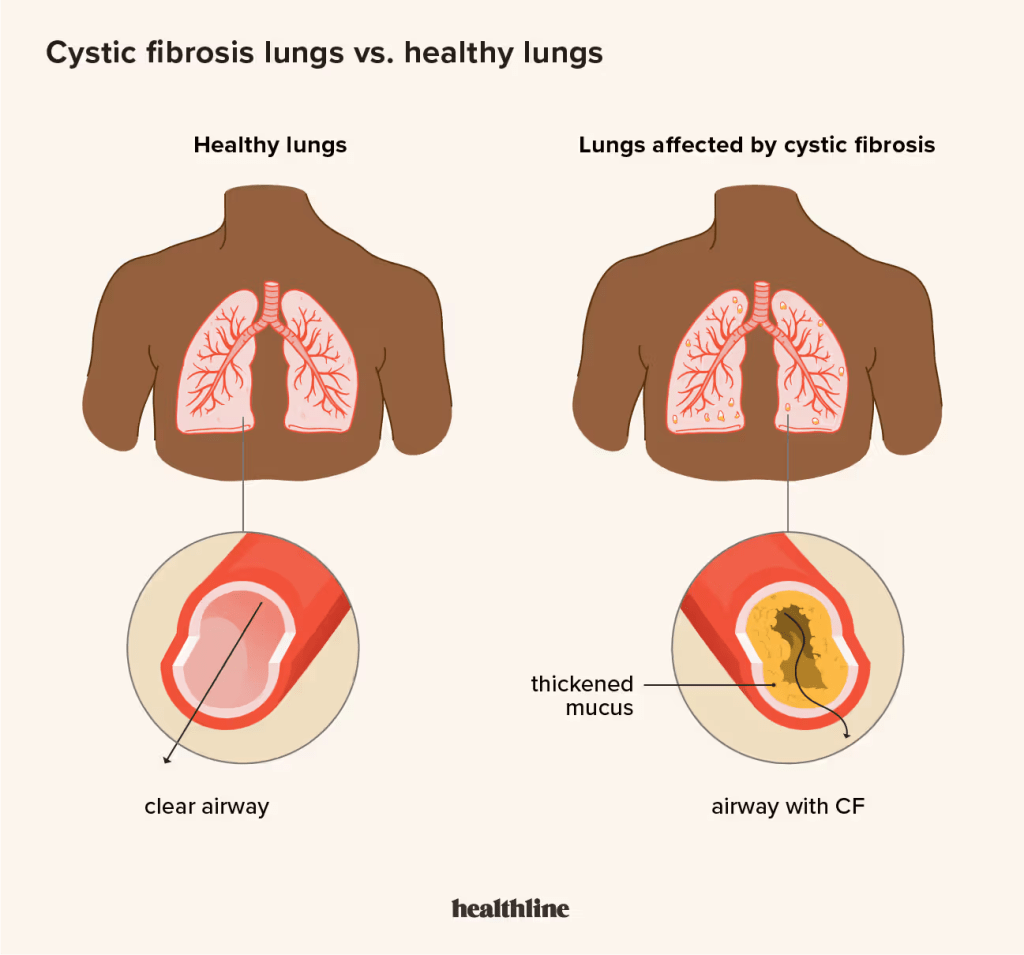
CF is a genetic disorder, inherited in an autosomal recessive manner. It’s caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which affects how water and salt flow and distribute themselves in and out of cells. CF affects the endocrine glands, resulting in the body’s production of unusually thick mucus that builds up in the lungs and intestines. This can cause serious inflammation and recurring infections. Since CF affects multiple organ systems, it causes a slew of symptoms and side effects, including chronic cough, shortness of breath, lung infections, malnutrition/poor growth, difficulty with bowel movements/constipation, and salty-tasting skin.
The resulting lung infections caused by CF tend to lead to more serious, life-threatening issues down the line. These issues include lung damage, scarred airways, lost lung functions, and respiratory failure. However, identifying the disease-causing gene has allowed researchers to begin to develop drugs to treat CF. These drugs increase the concentration of the protein produced the the gene. These drugs are specifically catered to CF, and most people (90%) with the disease are responsive to the treatment. Additional newborn screening programs in the United States have helped to reduce the number of people who develop the disease later on in their lives.
Symptoms:
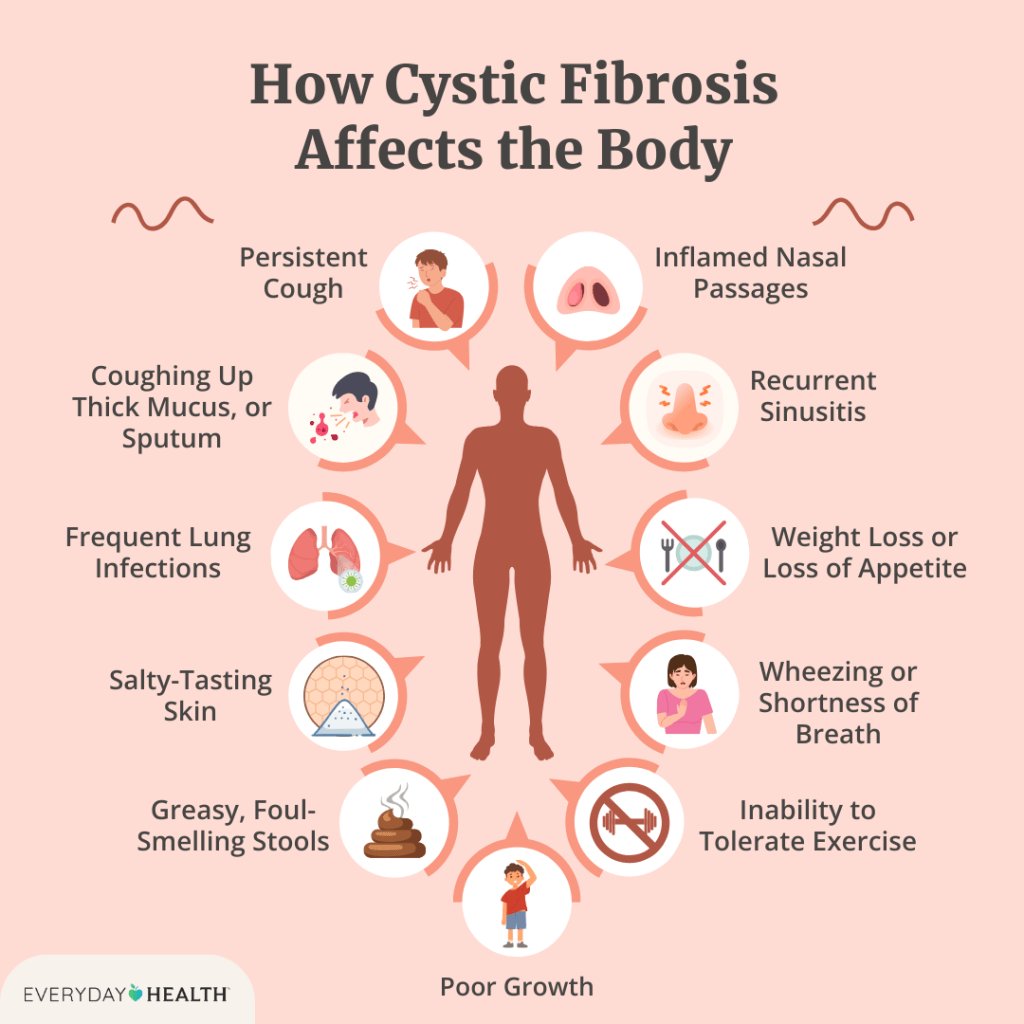
The symptoms of CF vary between individuals, causing some to only have minor respiratory problems, while others may have severe complications affecting several different organs throughout their body. The main symptoms include respiratory problems, pancreatic problems, gastrointestinal problems, and more.
Respiratory problems
Respiratory problems include difficulty breathing, wheezing, shortness of breath, chronic respiratory infections, labored breathing, nasal polyps, coughing up blood, lung collapse, and abnormally fast breathing.
Pancreatic problems
Pancreatic problems include mucus blocking intestinal digestive enzymes, malabsorption leading to insufficient nutrients, which causes poor growth/weight gain and nutritional deficiencies, especially of vitamins
Gastrointestinal problems
Gastrointestinal problems include Meconium ileus, which is when a newborn’s first stool is thick and blocks the intestines, constipation, abdominal pain, intestinal obstruction syndrome, stomach aches, cramping, nausea, vomiting, loss of appetite, and watery stools.
Additional symptoms
Additional symptoms include bloating, heartburn caused by gastroesophageal reflux, rectal prolapse, gallstones, and other gallbladder issues. Additional hormonal problems can arise which can cause diabetes, osteopenia, and osteoporosis. Problems with the exocrine system can cause the sweat glands to release extra salt that cannot be reabsorbed, leading to salty sweat, dehydration, abnormal heart rhythms, and overheating, especially due to physical activity. Musculoskeletal issues caused by CF include clubbing, arthritis, thoracic kyphosis, and scoliosis. Genital and urinary problems can also result from CF, including congenital bilateral absence of the Vas Deferens, low sperm count/sperm motility in men, and irregular ovulation and cervical mucus in women.
Causes:

CF is the result of a mutation to the CFTR gene, which is expressed in the cell lining of the airways, gastrointestinal tract, pancreas, and sweat glands. The CFTR gene produces a protein responsible for transferring chloride and sodium across the membrane of the cells. Without sodium, cells are unable to make salt water, resulting in the increased production of mucus in the cells. This mucus can cause tissue damage and inflammation. Currently, there are approximately 700 known mutations of the CFTR gene that cause CF, each of which results in a different level of protein synthesis or dysfunction, resulting in varying degrees of symptoms. The most common mutations are placed in six classes. Classes I, II, and III cause little to no protein function and result in a more severe version of the disease, but classes IV, V, and VI maintain some level of protein function, resulting in milder symptoms.
Additionally, the CFTR gene is inherited in an autosomal recessive manner, meaning that for an individual to develop CF, they must have two copies of the recessive allele that codes for a CF. However, if an individual inherits one CFTR allele and one normal allele, they will be a carrier, meaning that they will not show symptoms, but they may pass on the disease to their offspring. For each pregnancy where both parents are carriers of the disease, there is a 25% chance their child will have the disease, a 25% chance their child will be genotypically normal, and a 50% chance they will be carriers like their parents.
Diagnosis:

In the United States CF screening is standard soon after birth, in addition to physicians identifying the characteristic symptoms in a child, as well as looking through family history to see if there is evidence of a family member having had CF. Most of the U.S. conducts immunoreactive trypsinogen (IRT) assays on dried blood spots from newborns. This test helps to measure the amount of trypsinogen present in the newborn’s blood. Trypsinogen is formed by the pancreas causing high IRT levels in those with CF since the function of the pancreas may be hindered. Those who receive a positive IRT test should receive follow-up exams such as sweat testing or genetic testing. Sweat testing is a procedure in which the salt levels in one’s sweat are measured. Additionally, elevated chloride levels are cause diagnosis of CF, and intermediate chloride levels call for nasal potential difference testing (NPD). An NPD test tests how well nasal cells are able to move ions, which is hindered in individuals with CF as a result of faulty proteins meant to regulate ion movement. Post-diagnosis the parents and close relatives of the affected individuals should undergo genetic testing to confirm if they are carriers for the disease with the CFTR variant.
Treatment:
CF requires individualized care to make an impact on the symptoms expressed throughout the body. There is currently no cure for CF, but treatment is geared toward reducing the build up of mucus in the lungs to help prevent infections or any other conditions as a result of the mucus.
Lung therapies
Lung therapies include airway clearance methods to clear mucus from the lungs, chest physiotherapy to move mucus, and mechanical oscillating devices to loosen mucus.
Mucus thinners
Mucus thinners include dornase alfa, which is an aerosol medicine that can be inhaled from a nebulizer, helping to thin mucus. Mannitol is another aerosol mucus thinner; it helps to hydrate the airways and clear out mucus.
Other treatments
Other treatments include anti-inflammatory drugs and breathing support medicines. Lung transplantation may be necessary if damage to the lungs becomes beyond repair. Specific specialists may be required to help manage an individual’s symptoms.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop cystic fibrosis. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about cystic fibrosis, donation opportunities, or the progress being made on potential treatments, visit the Cystic Fibrosis Foundation! The Cystic Fibrosis Foundation strives to “to cure cystic fibrosis and to provide all people with CF the opportunity to lead long, fulfilling lives by funding research and drug development, partnering with the CF community, and advancing high-quality, specialized care.”
Let’s keep raising awareness! – Lily
References:
Thorne, A., Murphy, B., Calhoun, B., & Flume, P. A. (2025, January 17). Cystic Fibrosis Treatment, Symptoms, & Diagnosis | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/cystic-fibrosis/

Leave a comment