What is Gaucher Disease?:

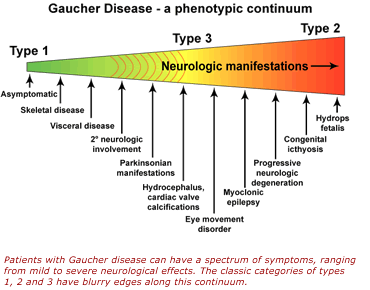
Gaucher Disease (GD) is a lysosomal storage disorder that causes an enlargement of the liver and/or spleen (hepatosplenomegaly), anemia, low platelet count (thrombocytopenia), and skeletal issues. In certain forms, it also affects the nervous system. The condition is caused by mutations in the GBA1 gene, leading to a deficiency in the enzyme glucocerebrosidase. This deficiency results in the buildup of glucosylceramide (Gb1) and its derivative glucosylsphingosine (Lyso-Gb1) in lysosomes, impacting various organs and systems. GD is classified into three types: non-neuronopathic (GD1), acute neuronopathic (GD2), and chronic neuronopathic (GD3). The symptoms of the disease vary based on the type, with GD1 appearing from early childhood to adulthood, GD2 manifesting before birth or shortly after, and GD3 symptoms typically emerging in early childhood. GD3 is characterized by gradually developing neurological symptoms such as cognitive decline, ataxia (lack of coordination), and myoclonic seizures, though the progression can differ widely. The disease’s impact on cognition can vary, with some patients showing only eye movement abnormalities. GD3 has three subtypes: GD3a, associated with myoclonic epilepsy; GD3b, which involves severe visceral issues like hepatosplenomegaly, growth delay, and skeletal abnormalities, but with milder neurological symptoms; and GD3c, or the cardiac type, which is linked to heart valve abnormalities and other cardiovascular complications.
Symptoms:

Individuals with Gaucher disease may experience a range of symptoms that vary in severity. One of the earliest signs is an enlarged spleen (splenomegaly), often leading to hypersplenism, where the spleen destroys blood cells, exacerbating anemia, thrombocytopenia, and leukopenia. Splenic infarcts, though rare, may cause abdominal pain and digestive problems. Liver enlargement (hepatomegaly) may also occur, often alongside splenomegaly, potentially leading to liver dysfunction. Bone abnormalities, such as bone pain, fractures, and skeletal deformities like Erlenmeyer flask deformities, are common, especially in childhood. Severe bone complications, such as osteonecrosis, can result in significant deformities and may require surgery or pain management. Hematological issues, including anemia and thrombocytopenia, can worsen over time, causing fatigue, weakness, and increased bruising or bleeding risk.
Neurological complications are typically seen in the more severe forms of the disease (GD2 and GD3) and include swallowing difficulties, seizures, muscle stiffness, coordination problems, and developmental delays. Additionally, individuals with Gaucher disease, particularly those with GD1 or carriers of GBA1 gene variants, have a higher risk of developing Parkinson’s disease or related neurodegenerative disorders, which may cause tremors, rigidity, and cognitive decline. Pulmonary complications, including respiratory distress, can occur in severe forms of GD due to lung involvement or aspiration. Chronic fatigue, pain, and cardiovascular issues, such as heart valve abnormalities and aortic calcification, are also reported. Other co-morbidities may include malignancies (such as multiple myeloma, lymphoma, and leukemia), immune dysfunction, and metabolic problems like hyperlipidemia, diabetes, and protein-losing enteropathy. Additionally, localized masses of Gaucher cells, known as Gaucheromas, can mimic tumors and cause diagnostic challenges.
Causes:
Gaucher disease is caused by genetic mutations in the GBA1 gene, which result in a deficiency of the enzyme glucocerebrosidase. This deficiency leads to the buildup of glucosylceramide and glucosylsphingosine in the lysosomes of various cells. Lysosomes are responsible for breaking down nutrients, including complex carbohydrates and fats, but in Gaucher disease, the lack of the enzyme causes abnormal accumulation of sphingolipids, leading to the disease’s symptoms. The primary cells affected are immune cells called macrophages, which are part of the reticuloendothelial system. The buildup of lipids triggers immune responses that result in inflammation, cell death, and damage to various tissues, including neurons, contributing to the disease’s symptoms. Gaucher disease follows an autosomal recessive inheritance pattern, meaning an individual must inherit a disease-causing gene variant from both parents to be affected. If a person inherits one normal gene and one disease-causing gene, they are carriers but typically do not show symptoms. The probability of two carrier parents having an affected child is 25% with each pregnancy, and there is a 50% chance the child will be a carrier. There is also a 25% chance the child will inherit normal genes from both parents. This risk is the same for both males and females.
Diagnosis:
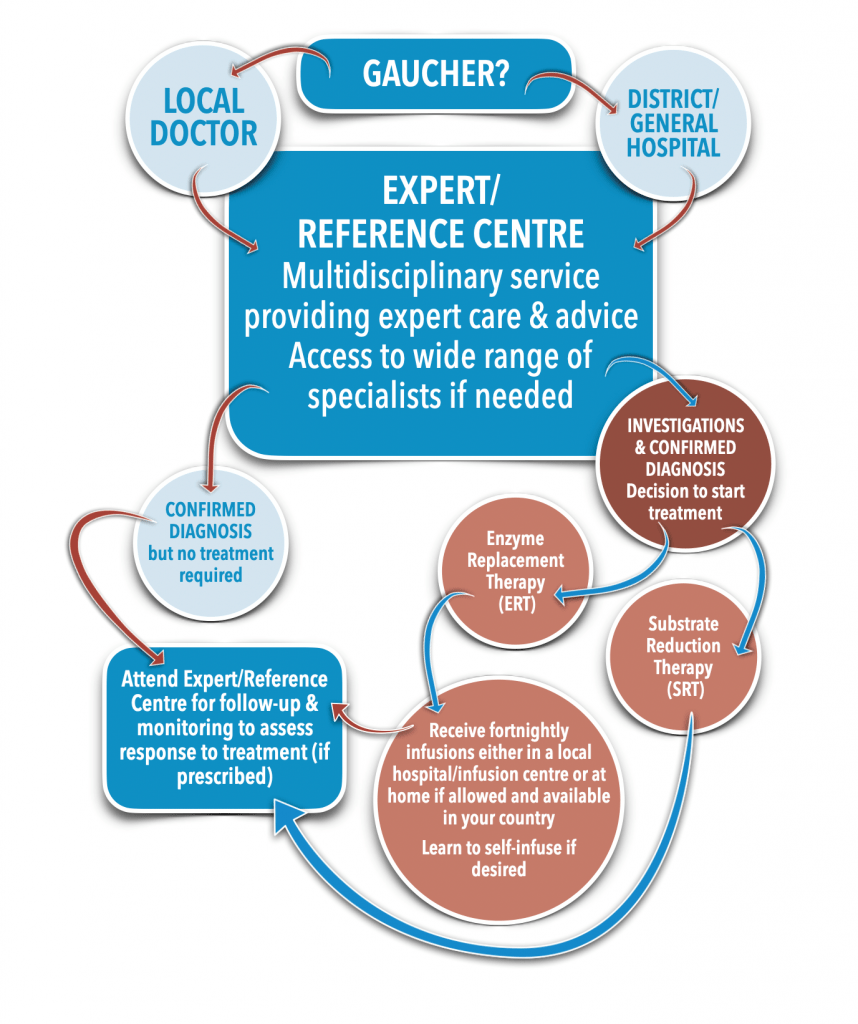
Gaucher disease may be suspected in individuals experiencing unexplained anemia, easy bruising, bleeding, enlarged spleen or liver, and chronic bone pain. The diagnosis is confirmed through an enzyme assay that measures acid beta-glucosidase activity in white blood cells or skin cells, along with genetic testing for variants in the GBA1 gene. Early diagnosis is particularly important for severe forms of the disease, as they progress quickly. Bone marrow examination may reveal Gaucher cells, which are enlarged macrophages with a distinctive “wrinkled tissue paper” appearance. While this can help confirm the diagnosis, it is not necessary when enzyme activity tests and genetic testing provide conclusive results. Other conditions, such as Niemann-Pick disease, chronic myeloid leukemia, and multiple myeloma, can also produce cells similar to Gaucher cells in the bone marrow, so these disorders must be considered in the diagnosis.
Treatment:
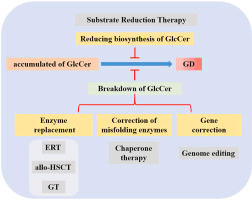
The primary treatment for Gaucher disease is Enzyme Replacement Therapy (ERT), which involves infusions of the recombinant enzyme glucocerebrosidase to help alleviate symptoms by reducing the accumulation of glucosylceramide. However, ERT does not significantly address brain-related issues due to the blood-brain barrier. Substrate Reduction Therapy (SRT) is another treatment option that reduces the production of glucosylceramide, and is used for patients with GD1 who cannot tolerate ERT or as an adjunct to it. Hematopoietic Stem Cell Transplantation (HSCT), which was used before ERT became available, has shown limited success and carries considerable risks. Symptom management focuses on pain relief and other supportive therapies, while regular monitoring is crucial to assess disease progression, treatment effectiveness, and potential complications. Additionally, supportive care, including nutritional guidance, physical therapy, and psychosocial support, plays an essential role in managing the condition and improving quality of life.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Gaucher disease. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about GD, donation opportunities, or the progress being made on potential treatments, visit the National Gaucher Foundation! The National Gaucher Foundation strives to “[serve] U.S. patients with Gaucher disease and their families. Through financial support, educational programming, patient services, and collaboration with medical professionals, NGF empowers Gaucher patients to live a better today.”
Let’s keep raising awareness! – Lily
References:
Goker-Alpan, O. (2024, September 9). Gaucher Disease – Symptoms, Causes, and Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/gaucher-disease/

Leave a comment