What is Phenykentonuria?:
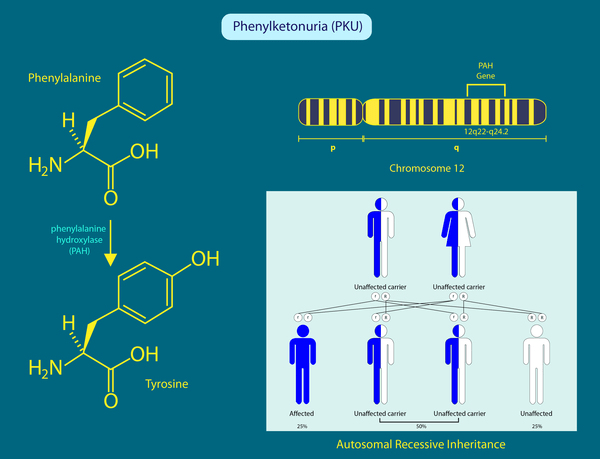
Phenylketonuria (PKU) is a genetic metabolic disorder that can be detected shortly after birth through routine newborn screening. It occurs due to a lack or deficiency of the enzyme phenylalanine hydroxylase (PAH), which is needed to break down the amino acid phenylalanine. Normally, PAH converts phenylalanine into tyrosine, but when this process is impaired, phenylalanine builds up to harmful levels in the brain. If untreated, this buildup can lead to severe intellectual disability. Early diagnosis and a strict low-phenylalanine diet starting in infancy can help prevent these complications.
Symptoms:
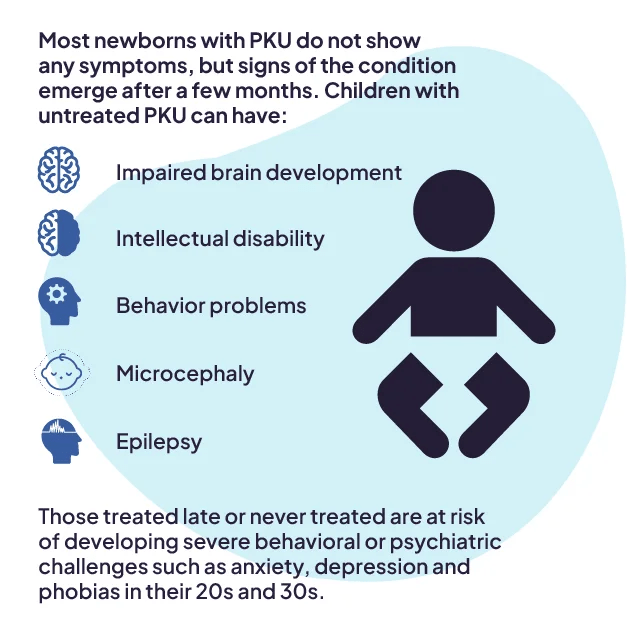
Babies with phenylketonuria (PKU) usually look healthy at birth. If diagnosed early and treated with a special diet, they may never show symptoms. However, if not treated soon after birth, symptoms can appear such as poor feeding, weakness, vomiting, irritability, and skin rashes. As they grow, untreated children often experience developmental delays and intellectual disability, with average IQs below 50. This brain damage is caused by high phenylalanine levels, which harm the protective myelin around nerves and reduce important brain chemicals like dopamine and serotonin.
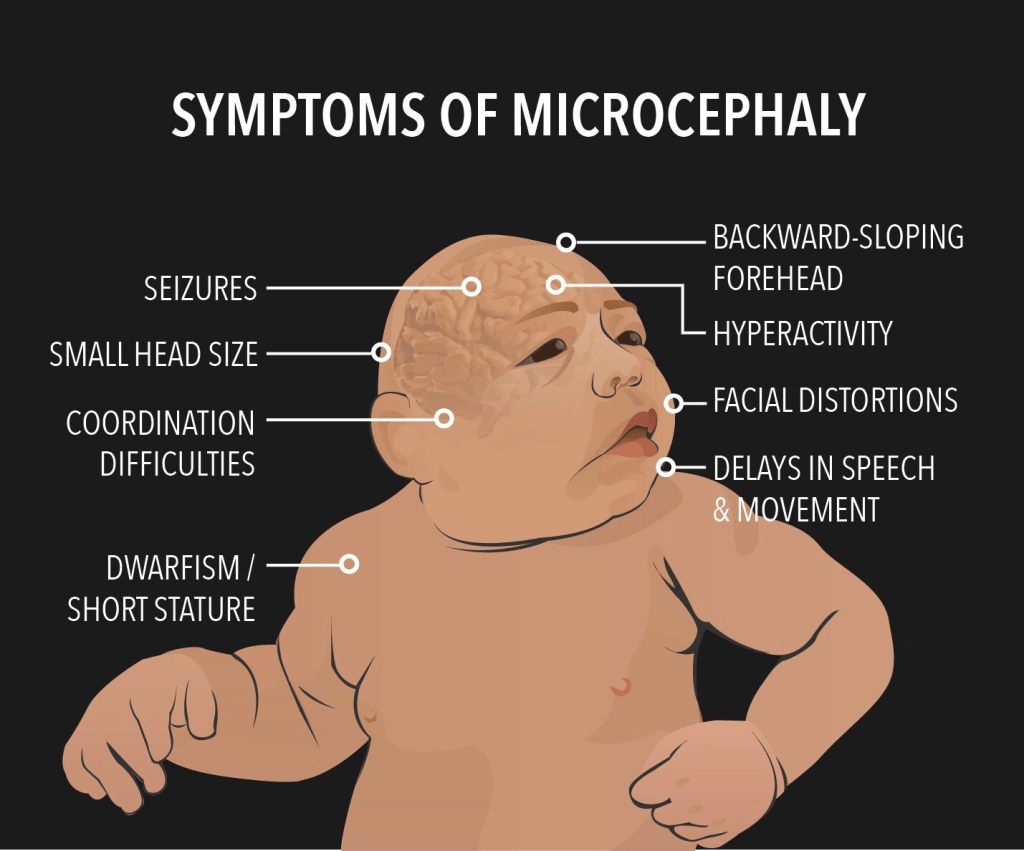
Untreated PKU can also affect appearance—infants may have lighter skin, hair, and eyes due to disrupted melanin production, and a distinct musty body odor caused by chemical buildup. Neurological issues like seizures, stiff or abnormal muscle movements, and tremors can also occur.
Women with untreated PKU face serious pregnancy risks, including miscarriage and birth defects in their children, such as heart problems, small head size, and facial abnormalities. These complications are linked to high phenylalanine levels, so it’s crucial for women with PKU to restart treatment before becoming pregnant and stay on it during pregnancy under medical supervision.
Causes:
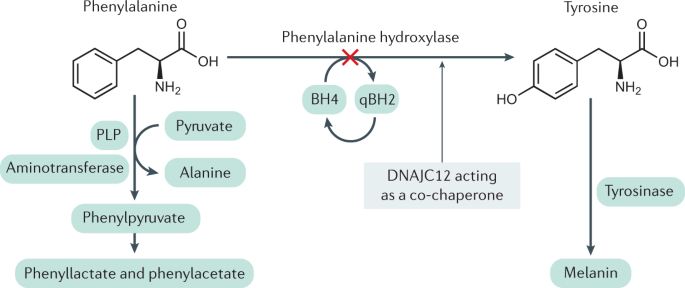
PKU is caused by mutations in the PAH gene, and over 300 different harmful variants have been found. These genetic differences affect how much PAH enzyme activity a person has, leading to different levels of phenylalanine buildup in the blood. Because of this, each person with PKU needs a diet tailored to their unique tolerance for phenylalanine.
The disorder is inherited in an autosomal recessive way, meaning a child must inherit one defective PAH gene from each parent to develop PKU. If both parents are carriers, there’s a 25% chance with each pregnancy that their child will have PKU, a 50% chance the child will be a carrier, and a 25% chance the child will inherit two normal genes. These odds are the same for both boys and girls.
Diagnosis:
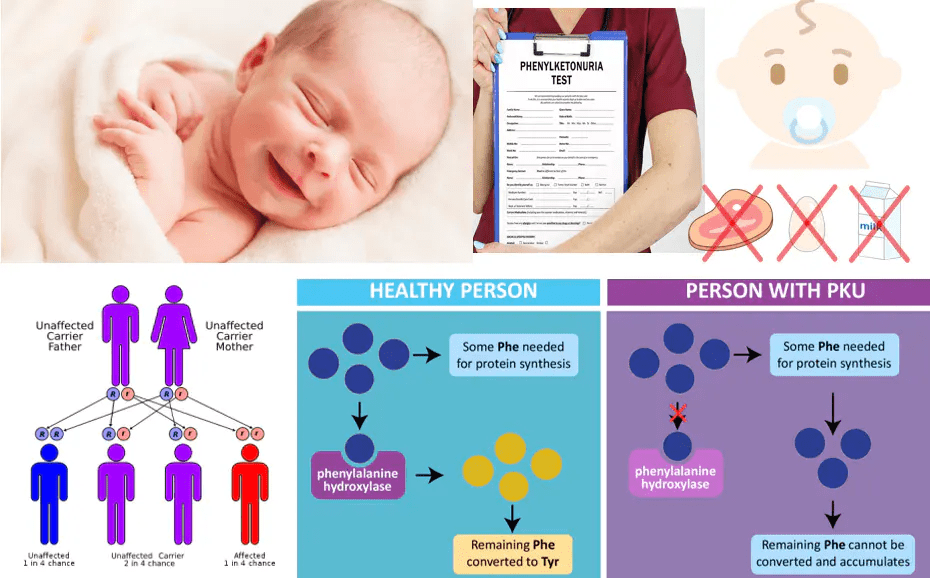
PKU is usually detected through standard newborn screening, which checks for high levels of phenylalanine in the blood—specifically, levels above 1,200 μmol/L (or 20 mg/dL). It can also be confirmed through genetic testing that identifies two harmful mutations in the PAH gene.
Treatment:
The main goal of PKU treatment is to maintain blood phenylalanine levels between 120–360 μmol/L (2–6 mg/dL). This is achieved through a carefully controlled diet, since phenylalanine is essential but harmful in excess. A strict low-phenylalanine diet, started early in life, can prevent intellectual disabilities and other complications like neurological, behavioral, and skin problems. Children treated before three months of age often develop normally, with IQs in the average range.
If someone with PKU stops managing their diet, they may experience cognitive decline and issues like mood swings, memory loss, poor concentration, depression, and coordination problems. As a result, it’s now widely accepted that the PKU diet should be lifelong. Many adults who restarted the diet after stopping it in childhood have seen improved mental function.
Because phenylalanine is found in nearly all natural proteins, dietary control requires special low-protein and phenylalanine-free foods. High-protein items like meat, dairy, and fish are avoided, while low-protein foods such as fruits and vegetables are allowed in limited amounts.
In terms of medication, the FDA approved Kuvan in 2007. This drug helps stimulate the remaining PAH enzyme activity and is used alongside the diet. In 2018, the FDA also approved Palynziq, an injectable enzyme therapy for adults whose phenylalanine levels remain too high despite diet and other treatments.
How You Can Make a Difference:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop PKU. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about PKU, donation opportunities, or the progress being made on potential treatments, visit the National PKU Alliance! The National PKU Alliance strives to “improve the lives of individuals with PKU, pursue a cure by expanding research and provide education and support to individuals living with PKU and their caregivers.”
Let’s keep spreading awareness! – Lily
References:
Paldeep S. Atwal. (2024, July 30). Phenylketonuria – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/phenylketonuria/

Leave a comment