What is NF1?:
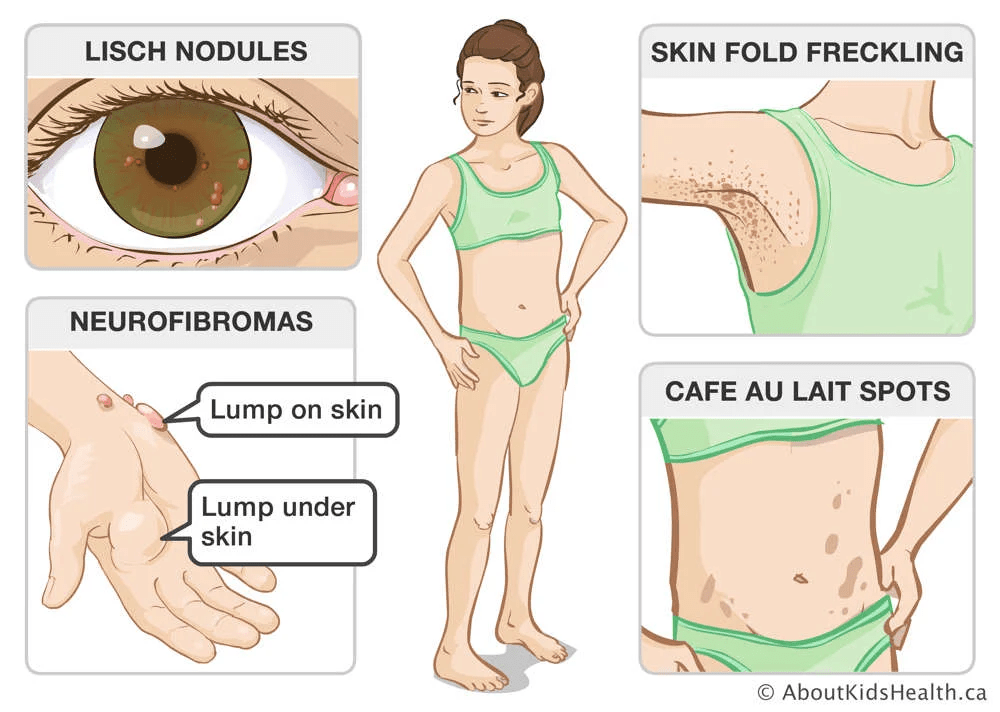
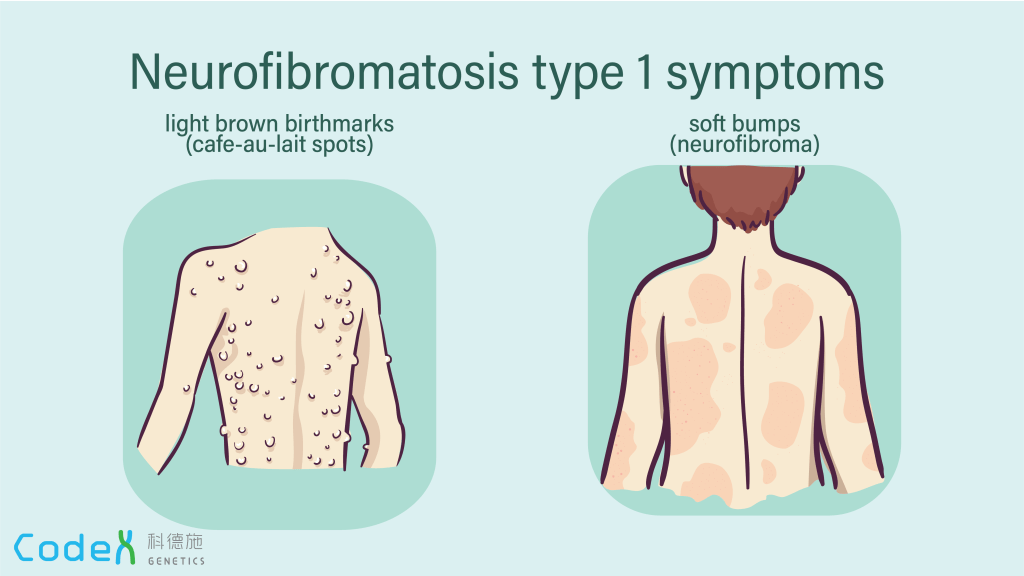
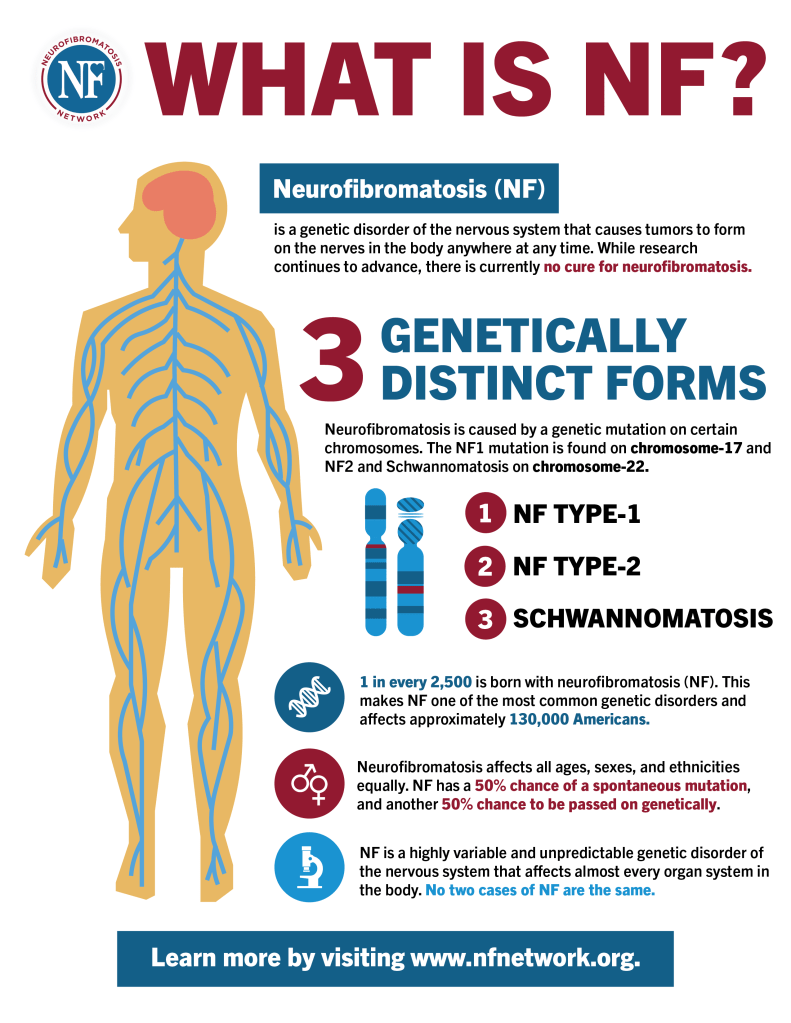
Neurofibromatosis type 1 (NF1) is a genetic condition marked by the development of both benign and malignant tumors, along with a range of skin, neurological, and developmental issues. The most common signs include nerve and skin tumors called neurofibromas and distinctive skin pigmentation changes, such as café-au-lait spots and unusual freckling in areas like the armpits and groin, typically visible within the first year of life.
Some individuals, even in early childhood, may develop larger, deep-seated benign tumors known as plexiform neurofibromas. Around half of NF1 patients have at least one of these. Additional features may include eye nodules, tumors along the optic nerves, and an increased risk for several types of cancer. Women with NF1, for instance, have a heightened risk of developing breast cancer in their 30s to 40s. A small but significant number also develop malignant peripheral nerve sheath tumors, often evolving from existing benign plexiform neurofibromas. Other tumors linked with NF1 include gastrointestinal stromal tumors, adrenal tumors, and gliomas in the brain or spine.
Physical characteristics often include a larger-than-average head, short stature, and skeletal issues like scoliosis, bowed legs, and bone malformations. Neurological challenges are common, including seizures, learning difficulties, attention problems, delayed development, and hyperactivity. Vascular problems, such as renal artery narrowing leading to high blood pressure, aneurysms, or even strokes, may also occur. NF1 is caused by mutations in the NF1 gene, which is responsible for producing neurofibromin—a protein that helps regulate cell growth and suppress tumors. The condition is inherited in an autosomal dominant manner, meaning a child has a 50% chance of inheriting it if one parent is affected. However, about half of cases arise spontaneously without a family history due to new mutations. NF1 tends to be highly variable in symptoms, even among individuals within the same family.
Symptoms:
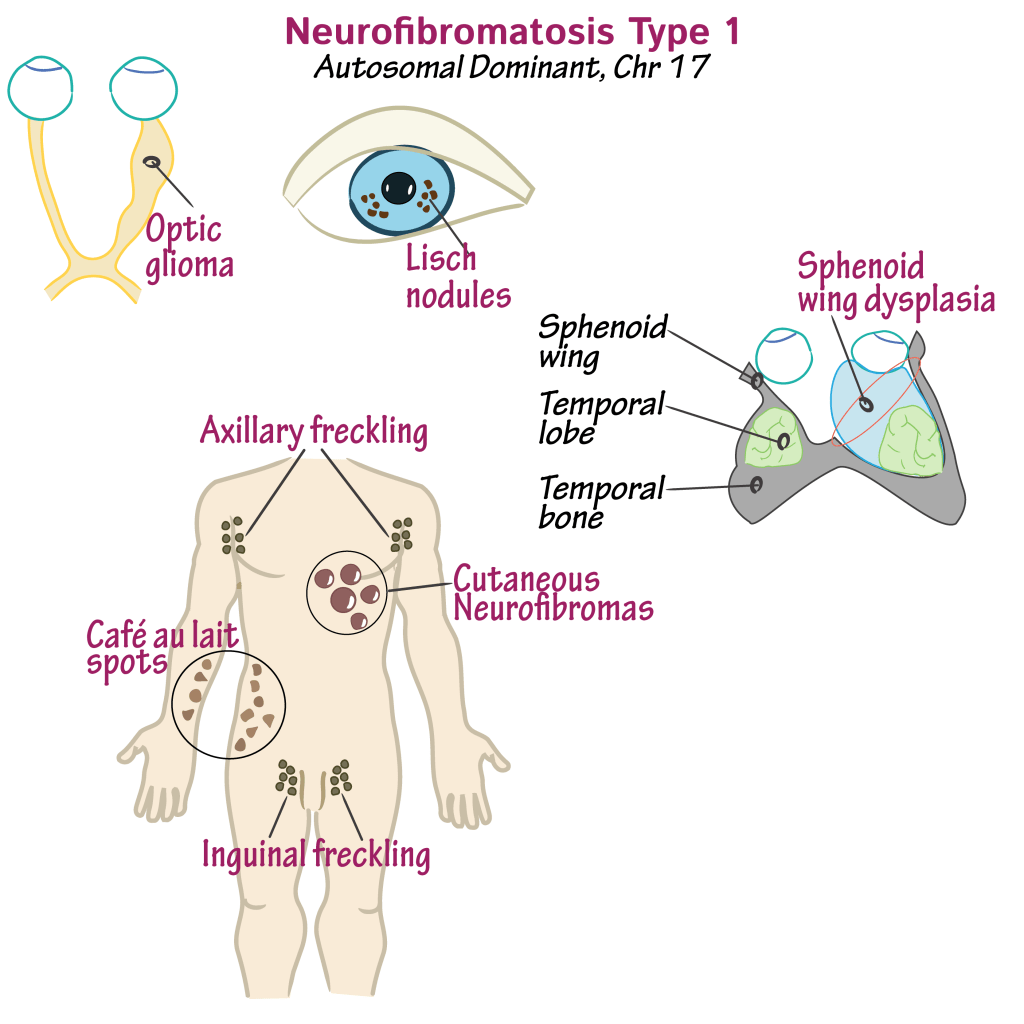
The diagnostic criteria for NF1 include six or more café-au-lait spots (5 mm before puberty or 15 mm after), freckling in the underarm or groin regions, two or more neurofibromas or one plexiform neurofibroma, optic pathway glioma, two or more eye nodules or choroidal abnormalities, specific bone deformities like sphenoid wing dysplasia or pseudoarthrosis, or a confirmed pathogenic NF1 gene variant. For individuals with a parent who has NF1, only one of these features is needed for diagnosis.
Symptoms typically appear in early childhood, and a diagnosis is often possible by age three. The condition progresses over time and may worsen during hormonal changes like puberty or pregnancy. NF1 presents very differently among individuals, even within the same family, and the severity and rate of progression are unpredictable. The earliest signs are usually café-au-lait macules and freckling in characteristic regions. Eye nodules, found in nearly all affected individuals, are also early and telling signs. Benign nerve tumors, called neurofibromas, can form under the skin or within organs and may lead to pain, disfigurement, or neurological dysfunction. These tumors increase in number with age and, in some cases, may transform into malignant peripheral nerve sheath tumors, which require immediate medical attention.
Around 15% of individuals with NF1 develop brain tumors (gliomas), particularly in the visual pathways, which can impair vision or trigger early puberty and abnormal head growth. Women with NF1 face a significantly increased risk of developing breast cancer, particularly between the ages of 30 and 40, and may also be at risk of a second cancer in the opposite breast after an initial diagnosis. NF1 is also associated with orthopedic complications such as scoliosis, bone fragility, and abnormal skull bone development. Bone density issues like osteopenia and osteoporosis are more prevalent, potentially linked to hormonal imbalances and increased bone breakdown. Individuals often have shorter stature, lower muscle strength, and larger head size than average.
High blood pressure is more common in people with NF1, often due to narrowing of the renal arteries or, more rarely, adrenal tumors like pheochromocytomas, which can dangerously elevate blood pressure. Sexual development may also be disrupted, with either delayed onset or early puberty, especially if associated with brain tumors affecting hormone regulation. Over half of affected individuals experience learning disabilities, often including ADHD, and may also suffer from seizures, headaches, or neurological symptoms such as numbness or weakness.
Causes:
NF1 is caused by harmful changes in the NF1 gene, which is responsible for producing neurofibromin—a protein that helps regulate cell growth by acting as a tumor suppressor. When this gene is altered, it leads to either reduced levels or malfunctioning neurofibromin, disrupting normal control of cell division and increasing the risk of tumor development. Many different mutations in the NF1 gene have been identified, but there is little correlation between a specific mutation and the severity or type of symptoms experienced. This variability, even among family members with the same genetic change, is thought to result from interactions with other genetic factors and environmental influences.
NF1 follows an autosomal dominant inheritance pattern, meaning only one copy of the altered gene—either from the mother or father—is enough to cause the disorder. About half of all NF1 cases are inherited from a parent with the condition. The other 50% result from new mutations in the NF1 gene that occur randomly in the affected individual without a family history.
In dominant genetic conditions like NF1, each child of an affected parent has a 50% chance of inheriting the disorder, regardless of sex. A more localized form of the disease, called segmental NF1, occurs when the mutation arises during early development, affecting only a portion of the body. This form is generally milder and restricted in distribution. Although segmental NF1 is usually not inherited, there is a potential—depending on how widespread the mutated cells are in the parent’s body—that the mutation could be passed on to offspring, possibly resulting in classic NF1. The exact risk of transmission in these cases is not fully known.
Diagnosis:
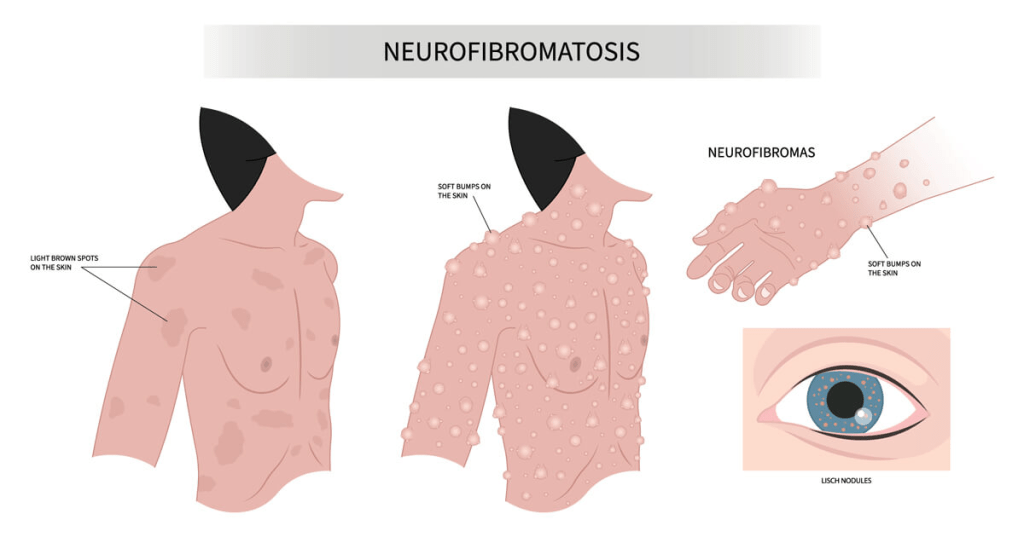
NF1 is typically diagnosed within the first ten years of life, often based on visible signs such as café-au-lait spots, freckling, optic gliomas, or bone abnormalities like pseudoarthrosis. If any one of these features is observed, NF1 should be considered. For individuals without an affected parent, a diagnosis requires at least two specific clinical or genetic features; for those with a parent who has NF1, only one of these findings is needed.
Diagnostic criteria include: six or more café-au-lait spots (≥5 mm before puberty or ≥15 mm after), freckling in the underarm or groin regions, two or more neurofibromas or one plexiform neurofibroma, optic pathway glioma, two or more Lisch nodules or choroidal abnormalities seen on specialized eye imaging, specific bone deformities such as sphenoid wing dysplasia or bowed long bones, or detection of a disease-causing NF1 gene variant in normal tissue, with the mutation present in about 50% of the gene copies. Diagnosis is primarily clinical, though genetic testing is increasingly used to confirm the condition or assist in reproductive decision-making. Some skin signs, like café-au-lait spots, may not be clearly visible and might require examination under ultraviolet light to be properly identified.
Treatment:
Management of NF1 typically involves regular monitoring, with treatment focused on specific symptoms or complications. For cutaneous, subcutaneous, and deep neurofibromas—which are usually benign—active surveillance is often preferred. However, surgery may be considered for tumors that cause pain, disfigurement, or functional issues. Smaller skin lesions might also be treated using laser or electrocautery techniques. While radiation and chemotherapy are occasionally used, their benefits are uncertain and often outweighed by risks; ongoing clinical trials aim to clarify their effectiveness.
Plexiform neurofibromas, which are typically large and deeply embedded, are rarely suitable for complete surgical removal. In recent years, targeted drug therapies have become available. Selumetinib and mirdametinib are FDA-approved medications for inoperable, symptomatic plexiform neurofibromas in children and adults as young as two. These drugs, known as kinase inhibitors, work by blocking a key enzyme that drives abnormal cell growth, and have shown promise in reducing tumor size and improving symptoms. Supportive treatments such as physical therapy and orthopedic devices can improve mobility and quality of life, especially for those with skeletal complications like scoliosis. Bracing or even surgery may be required in severe cases.
Ongoing surveillance is essential for NF1 management. Annual physical exams, blood pressure checks, and frequent eye exams are recommended for all patients. Children in particular should receive yearly vision screening, head circumference monitoring, and developmental assessments. Imaging techniques like MRI or PET scans may be used to monitor internal tumors, with whole-body MRI being studied as a comprehensive tool. For women with NF1, annual breast cancer screening should start at age 30 using mammography or MRI. Additional specialist care may be required to monitor or treat complications involving the nervous system, bones, blood vessels, or vision. Genetic counseling is strongly recommended for affected individuals and their families to understand the inheritance pattern, assess risks, and guide family planning.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop NF1. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about NF1, donation opportunities, or the progress being made on potential treatments, visit the Neurofibromatosis Network! The National PKU Alliance strives to “to find treatments and a cure for neurofibromatosis by promoting scientific research, improving clinical care, providing outreach through education and awareness, while offering hope and support to those affected by NF.”
Let’s keep spreading awareness! – Lily
References:
Zupan, M., Bundra, K., & Jordan, J. T. (2025, April 23). Neurofibromatosis 1 – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/neurofibromatosis-type-1-nf1/

Leave a comment