What is EDS?:
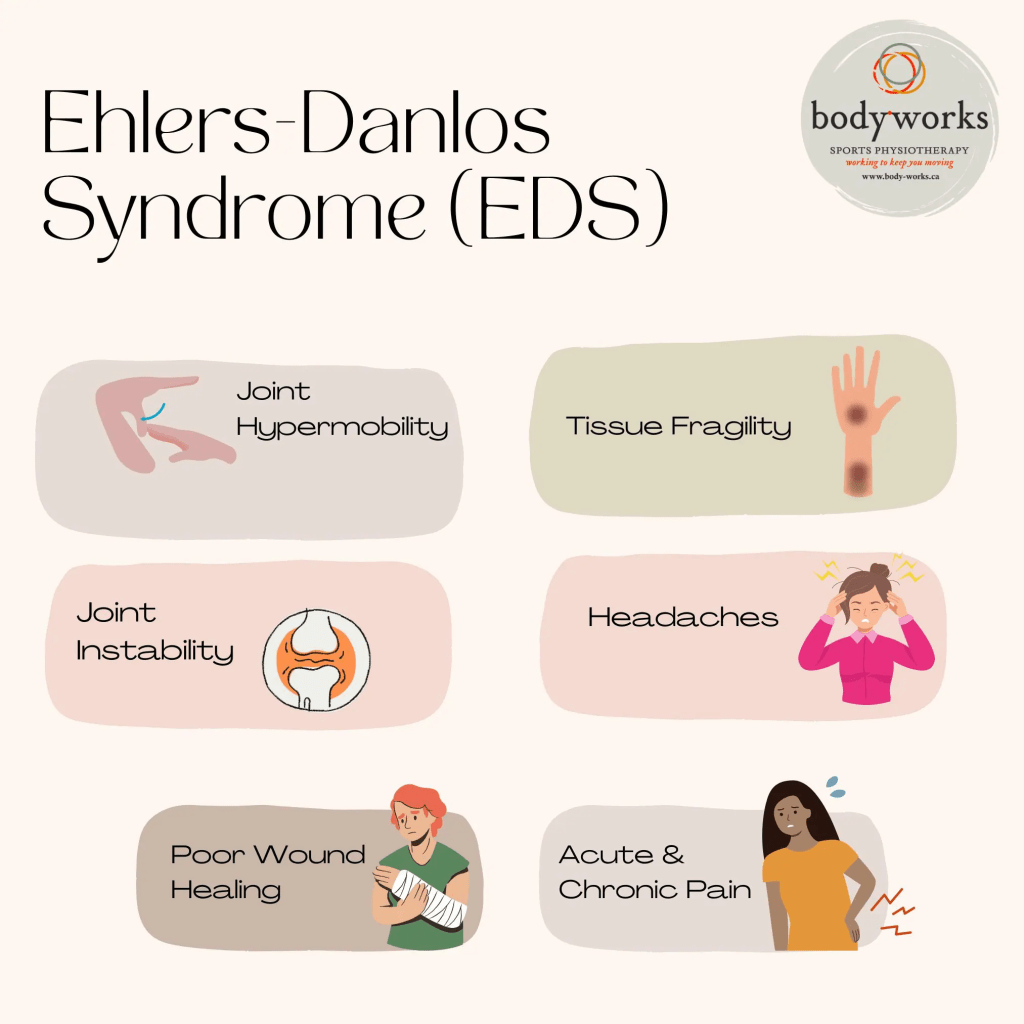
Ehlers-Danlos syndrome (EDS) is a group of genetic disorders that affect the body’s connective tissue due to abnormalities in collagen, a key structural protein. Collagen plays a vital role in strengthening tissues like bones and providing flexibility to tissues like cartilage. In EDS, the collagen may either be weak or insufficient in amount, leading to issues in the skin, joints, muscles, skeleton, and blood vessels. People with EDS often have soft, loose, and stretchy skin that heals poorly and forms very thin scars. Their joints tend to be overly flexible (hypermobile), making them prone to frequent dislocations. Fragile blood vessels also make bruising and serious bleeding more likely.
Each subtype of EDS stems from a unique genetic mutation, and while individuals within a subtype share core features, symptoms can still vary widely. The classification of EDS has evolved over time—from an initial system of Roman numerals, to six types based on clinical traits, and most recently, to a 2017 system recognizing 13 subtypes. This latest classification also includes a research-based grouping (Groups A–F) focused on genetic differences.
Symptoms:

Classical Ehlers-Danlos Syndrome (cEDS), previously known as EDS types I and II, primarily involves stretchy skin, joint looseness, and fragile blood vessels. A key feature is the development of very thin, discolored scars that stretch over time, especially in areas prone to pressure like the knees, elbows, shins, and forehead. These scars often appear papery in texture.
Joint dislocations or partial dislocations are common but typically manageable. Other possible signs include soft, fleshy skin growths called molluscoid pseudotumors and firm, movable lumps under the skin known as calcified spheroids.
Some individuals with cEDS may also have problems with heart valves, particularly the mitral valve. Weakness in these valves can lead to blood leaking backward, which may strain the heart over time and lead to congestive heart failure. Additionally, there is a heightened risk of aortic complications, such as dilation or dissection. Aortic dissection, a tear between the layers of the aorta, can reduce blood flow to vital organs and is a medical emergency. Sudden, severe chest pain in cEDS patients requires immediate medical attention.
Causes:

Ehlers-Danlos Syndrome (EDS) can be inherited in either a dominant or recessive pattern. Humans inherit two copies of each gene, one from each parent. In dominant inheritance, only one faulty gene copy is needed to cause the disorder. This gene may be inherited from a parent or may arise from a new mutation. Each child of an affected parent has a 50% chance of inheriting the condition, regardless of gender. In some cases, the mutation appears spontaneously and is not inherited.
In recessive inheritance, a person must inherit two abnormal gene copies to develop the condition. Carriers, who have one mutated and one normal gene, typically show no symptoms. If both parents are carriers, there’s a 25% chance their child will have EDS, a 50% chance the child will be a carrier, and a 25% chance the child will inherit two normal genes. When someone is diagnosed with EDS, genetic counseling and testing are recommended to understand the inheritance pattern within the family.
Different EDS subtypes are linked to specific genes involved in producing or processing collagen, the main structural protein in connective tissues. These genes include COL1A1, COL1A2, COL1A3, COL5A1, COL5A2 (collagen production) and ADAMTS2, PLOD1, TNXB (collagen processing).
For the classical type, the condition follows an autosomal dominant pattern involving mutations in the COL5A1 and COL5A2 genes. These genes code for precursor proteins like pro-alpha1(V) and pro-alpha2(V), which are processed into mature collagen. This type V collagen then combines with type I collagen to help regulate the size and structure of collagen fibrils, ensuring proper connective tissue function.
Diagnosis:
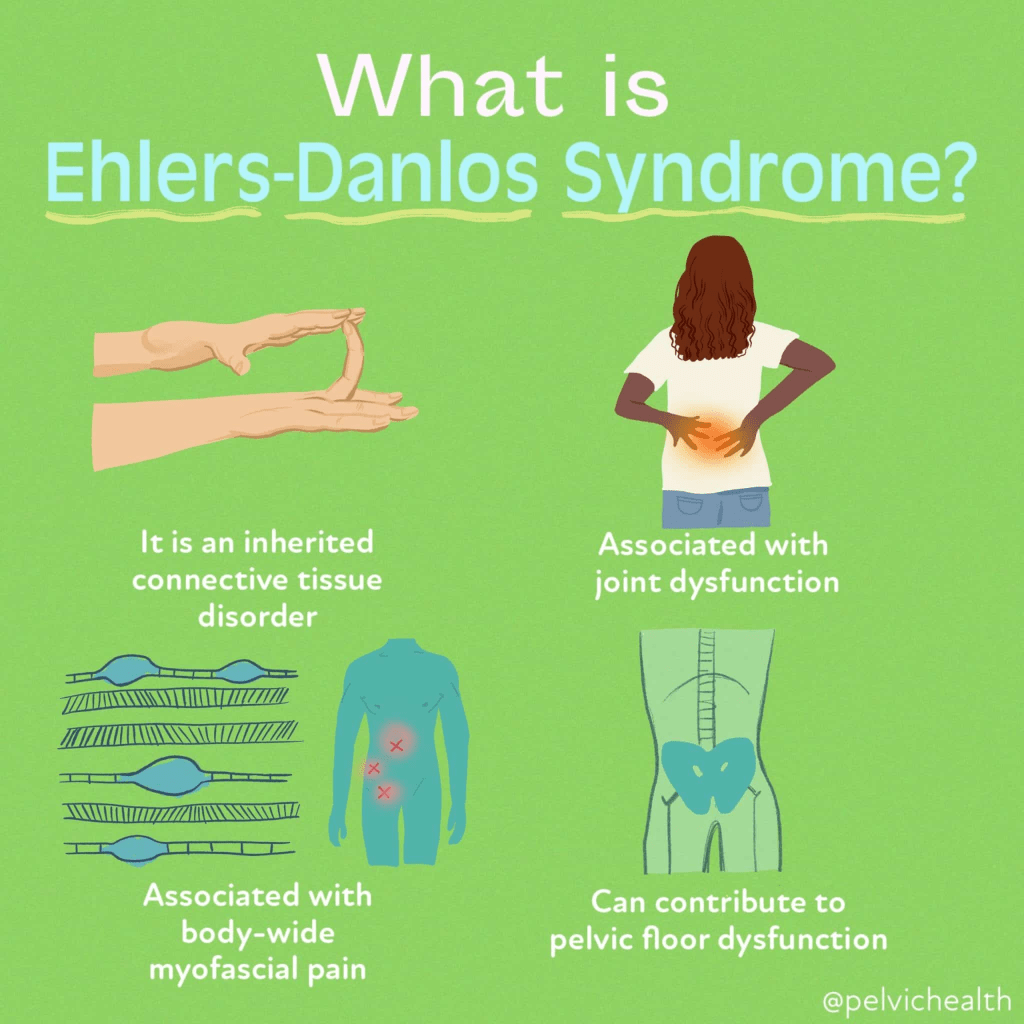
Ehlers-Danlos Syndrome (EDS) is generally diagnosed through a combination of clinical evaluation and patient history. Physicians look for characteristic features such as skin hyperextensibility and joint hypermobility. Skin stretchiness is measured by gently pulling the skin at a neutral site until resistance is felt, while joint flexibility is typically assessed using tools like the Beighton score. In many cases, genetic testing can help confirm a diagnosis by identifying specific gene mutations linked to various EDS subtypes. For example, mutations in the COL5A1 gene can confirm classical EDS (cEDS). However, in hypermobile EDS (hEDS), no specific gene has yet been identified, so diagnosis is made by ruling out other known types through genetic testing.
Additional diagnostic tools include imaging studies such as CT scans, MRIs, and echocardiograms. These are used to detect complications like mitral valve prolapse or aortic dilation, which are sometimes associated with EDS. CT scans provide cross-sectional images using X-rays, MRIs use magnetic fields to image soft tissues and organs, and echocardiograms use sound waves to examine heart structure and function. In some cases, specialized X-rays are used to detect calcified spheroids (firm lumps under the skin), assess spinal abnormalities such as scoliosis or kyphosis, or measure bone density in cases of suspected osteopenia, especially relevant in rarer forms like kyphoscoliotic or arthrochalasia EDS.
Electron microscopy of tissue samples may also reveal distinct collagen abnormalities that support an EDS diagnosis. For certain subtypes like kyphoscoliotic EDS (kEDS), laboratory tests can further aid diagnosis by analyzing enzyme activity in skin fibroblasts or by measuring specific biochemical markers in urine, such as the ratio of deoxypyridinoline to pyridinoline cross-links. Altogether, a thorough combination of clinical, genetic, imaging, and laboratory evaluations is essential for an accurate diagnosis of EDS and its various subtypes.
Treatment:
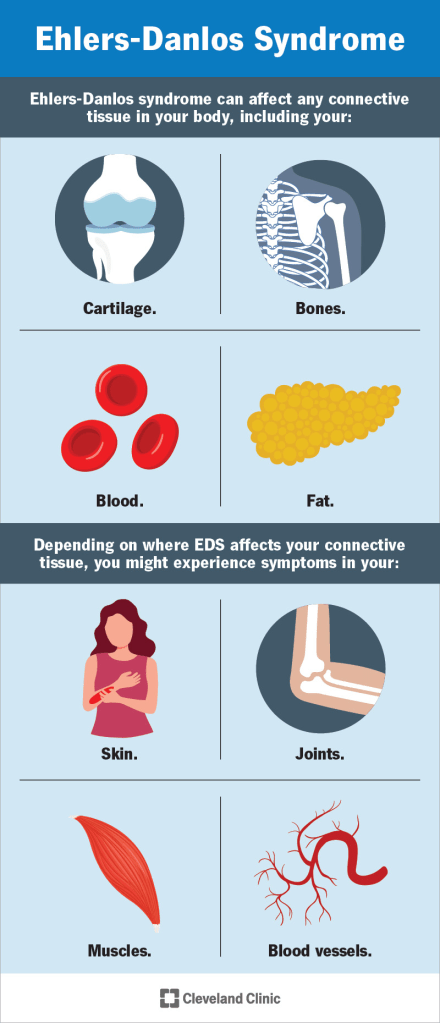
The management of Ehlers-Danlos Syndrome (EDS) primarily focuses on preventing serious or life-threatening complications, especially those affecting the skin, joints, and cardiovascular system. Due to fragile, stretchable, and thin skin, wound healing can be problematic. In cases of injury or surgery, doctors use deep sutures generously and carefully place superficial ones to reduce scarring. Stitches are often left in longer than usual to support proper scar formation. Vitamin C (ascorbic acid) may also be recommended to help minimize easy bruising, a common symptom in EDS.
Joint instability is another key concern, as hypermobile joints dislocate easily, and repeated dislocations become more likely over time. To avoid injury, patients are advised to limit strenuous activities, heavy lifting, and contact sports. Vascular fragility poses serious risks for internal bleeding and arterial dissection. Managing blood pressure is critical, as hypertension increases strain on fragile vessels. Non-invasive imaging techniques such as ultrasound, MRI, or CT scans are preferred for vascular monitoring, while invasive procedures like arteriography or colonoscopy should be used cautiously and only when clearly beneficial. Elective surgeries and non-urgent procedures should be carefully weighed against potential risks. Pregnancies in EDS patients require close monitoring by high-risk obstetric specialists, as labor can progress rapidly and delivery methods should be individualized. For those with aortic root dilation, echocardiograms should be performed each trimester. All EDS patients should seek immediate care for sudden pain and are encouraged to wear a MedicAlert bracelet in case of emergencies.
Patients with hypermobile EDS (hEDS) may benefit from physical therapy, low-impact exercise, and supportive devices such as braces, wheelchairs, or ergonomic tools. Special mattresses and adaptive writing utensils can help reduce musculoskeletal pain. Pain management is tailored to each person, and gastrointestinal or psychological issues are addressed based on individual needs. Bone health is also important; calcium, vitamin D, and biennial DEXA scans can help maintain bone density. Those with kyphoscoliotic EDS (kEDS) should have regular eye exams due to risks of globe rupture, retinal detachment, and glaucoma. Patients with dermatosparaxis EDS (dEDS) may need protective coverings for vulnerable areas like elbows and knees to prevent skin injury.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop EDS. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about EDS, donation opportunities, or the progress being made on potential treatments, visit the Ehlers-Danlos Society! The Ehlers-Danlos Society is “dedicated to advancing and accelerating research and education in Ehlers-Danlos syndromes (EDS) and hypermobility spectrum disorders (HSD). We support the development of effective and equitable EDS and HSD therapies and work collaboratively to improve the lives of individuals affected by EDS and HSD.”
Let’s keep spreading awareness! – Lily
References:
Chepa-Lotrea, X., & Francomano, C. (2021, September 27). Ehlers Danlos Syndrome – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/ehlers-danlos-syndrome/

Leave a comment