What is Angelman Syndrome?:
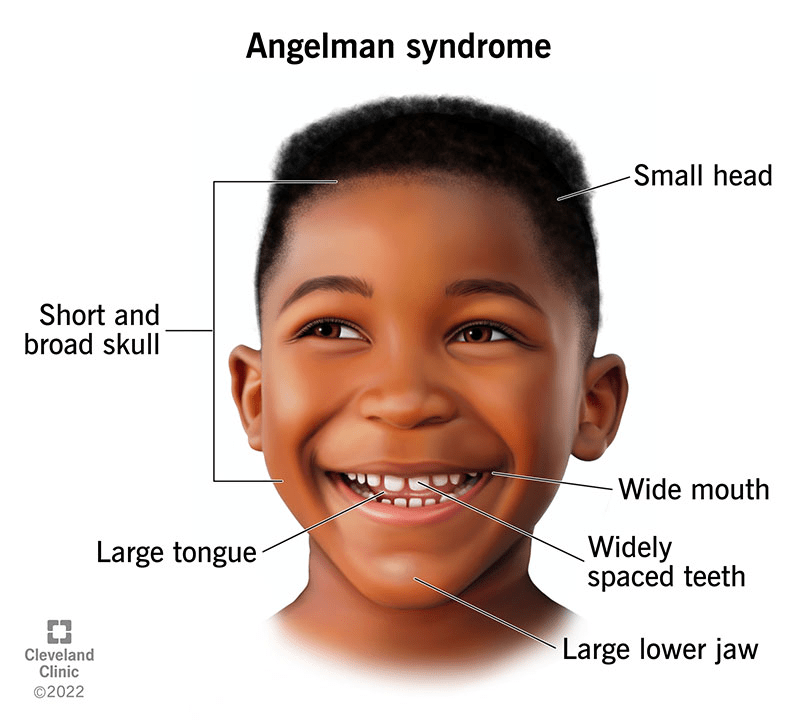
Angelman syndrome is a rare genetic and neurological condition primarily marked by significant developmental delays, severe learning difficulties, and minimal or absent speech. Affected individuals often have trouble with voluntary movement (ataxia), experience tremors, and exhibit jerky limb movements. A notable behavioral trait is a consistently happy demeanor, including frequent, spontaneous smiling and laughter.
While most individuals are unable to speak, many learn alternative ways to communicate, such as through gestures. Their ability to understand language (receptive language) is often better than their ability to express it. Additional symptoms can include seizures, sleep disturbances, and feeding problems. Although some children may show distinctive facial traits, these usually resemble those of their parents. The disorder results from a deletion or malfunction of the UBE3A gene. First identified by Dr. Harry Angelman in 1965, the condition typically isn’t recognized at birth. Diagnosis is most often made between the ages of one and four years.
Symptoms:

Angelman syndrome presents a wide range of symptoms that differ from person to person. Not all individuals will experience the same features. Some may have seizures while others do not, and speech ability can vary, with most individuals unable to speak more than a few words, though some have limited speech. Children with Angelman syndrome typically face significant developmental delays and learning disabilities. Communication is often severely affected, with most children unable to speak fluently but able to understand simple instructions. Many eventually learn to communicate through gestures or using communication aids like boards.
Movement and coordination difficulties are early signs, often appearing between 6 and 12 months of age. These can include ataxia (poor coordination), jerky movements, and a distinctive posture where the arms are held up with bent wrists and elbows. Children may flap their hands, especially when excited. Low muscle tone in the torso, increased tone in the limbs, and exaggerated reflexes are also common. Motor development, such as walking, is delayed. Some walk between ages 2 and 3, others later, and around 10 percent may never walk unaided.
Behaviorally, individuals with Angelman syndrome typically have a consistently happy demeanor, frequent smiling, and bouts of unprovoked laughter. They are often hyperactive and highly excitable, constantly in motion and eager to explore their environment. Some individuals have smaller head sizes (microcephaly), and seizures are common, usually starting between ages 1 and 5 and often improving during adolescence. Facial features can include a wide mouth, prominent chin, deep-set eyes, protruding tongue, widely spaced teeth, and a flat back of the head, although these vary.
Feeding problems may occur during infancy, such as difficulty sucking or swallowing, though these are usually mild. Digestive issues like constipation and acid reflux (GERD) can also be present. Certain individuals show reduced pigmentation in their skin, eyes, and hair due to a lack of melanin, which may lead to light sensitivity, involuntary eye movements (nystagmus), and reduced visual clarity. Strabismus (crossed eyes) and excessive drooling are also reported.
Sleep disturbances are common, including reduced need for sleep and disrupted sleep patterns. Children may also have an unusual interest in water, music, or shiny objects and may be overly sensitive to heat. As they grow older, individuals with Angelman syndrome may develop spine curvature (scoliosis), joint stiffness, and decreased mobility. Facial features such as a more prominent lower jaw may become more noticeable in adults. Some may also develop eye conditions like keratoconus and may become overweight. Despite these challenges, puberty typically occurs normally, and fertility is possible in some cases.
Causes:
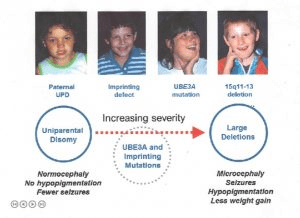
Angelman syndrome is caused by a deficiency in the expression of the UBE3A gene, which is located on the long arm of chromosome 15 at region 15q11-q13. This gene is essential for brain function, as it produces a protein involved in marking damaged or unnecessary proteins for breakdown (ubiquitination). In the brain, only the maternal copy of the UBE3A gene is active, while the paternal copy is typically silenced due to a genetic process called genomic imprinting. If the maternal copy is missing or inactive, the gene cannot function in the brain, resulting in the symptoms of Angelman syndrome.
There are several genetic mechanisms that can disrupt the normal expression of the UBE3A gene. In about 70–75% of cases, the cause is a microdeletion in the maternal chromosome 15 that includes the UBE3A gene. These deletions usually occur spontaneously and are not inherited, with a low recurrence risk of 1–2%. In approximately 3–5% of cases, the disorder is caused by an imprinting defect, where the maternal copy is incorrectly silenced, often due to abnormal DNA methylation. About 20% of these imprinting defects are caused by deletions in the Imprinting Center, while the rest are due to unknown causes. If the imprinting defect involves a deletion, the chance of recurrence can be as high as 50%.
Another 2–5% of cases are caused by paternal uniparental disomy, a rare event where both copies of chromosome 15 are inherited from the father, leaving no active maternal copy of UBE3A in the brain. This type also carries a very low recurrence risk. In 10–20% of cases, Angelman syndrome is the result of a mutation in the UBE3A gene itself. These mutations interfere with the gene’s function and can cause all core symptoms of the syndrome. If inherited, these mutations may also have a recurrence risk of up to 50%.
Rarely, Angelman syndrome may result from complex chromosomal rearrangements involving chromosome 15, which can increase the chance of recurrence in families. In about 10% of cases, no identifiable genetic cause is found. Some individuals in this group may have undetected mutations in UBE3A or defects in other, still-unknown genes that mimic the clinical features of Angelman syndrome.
Diagnosis:
The diagnosis of Angelman syndrome is typically based on a detailed medical history, clinical evaluation, and recognition of characteristic symptoms. Around 80% of cases can be confirmed through specialized blood tests. One common test is DNA methylation analysis, which can identify abnormalities in gene expression but cannot distinguish between the specific underlying causes such as chromosome deletions, imprinting defects, or paternal uniparental disomy. To detect the deletion of the 15q11-q13 region, which occurs in about 70% of cases, tests like fluorescent in situ hybridization (FISH) or, more commonly, chromosomal microarray analysis are used.
For individuals who do not show abnormalities on methylation testing, mutation analysis of the UBE3A gene can identify an additional 10% of cases. This analysis can be done as a targeted test for UBE3A or more frequently as part of broader genetic testing such as whole exome sequencing, which screens thousands of genes linked to intellectual disabilities and developmental disorders.
Treatment:

Currently, treatment for Angelman syndrome focuses on managing symptoms and providing supportive care, as no cure or gene-specific therapy is available yet. Although several clinical trials are underway, treatment options remain limited to addressing individual symptoms. However, ongoing advances in neuroscience and gene therapy offer promising future possibilities for more targeted and potentially curative treatments.
Overall, individuals with Angelman syndrome generally enjoy good physical health and can receive standard pediatric care, including routine vaccinations. Seizures, which are common in this condition, are typically managed with anti-seizure medications. While many individuals respond well to a single medication, some may require multiple drugs for effective control. No single anticonvulsant has proven universally effective. Sleep disorders are frequent and may be managed through behavioral strategies and consistent sleep routines; in some cases, sedative medications may also be used.
Feeding challenges, particularly in infancy, may be addressed with special feeding techniques or equipment like modified nipples. Gastroesophageal reflux can be treated with medications that support digestion or, in severe cases, surgery to tighten the esophageal valve. Constipation may be managed with laxatives. Mobility issues such as difficulty walking can be improved with physical therapy and the use of ankle braces. In about 10% of cases, scoliosis may develop and require bracing or surgical intervention. Strabismus (crossed eyes) might also require corrective surgery.
Early intervention is critical to help children with Angelman syndrome reach their full potential. Supportive services can include access to educational, medical, social, and vocational programs. Physical, speech, and occupational therapies are often beneficial. Behavioral therapy may help address problematic behaviors, and assistive communication technologies, such as voice-output devices or picture-based systems, can significantly improve social and learning outcomes. Finally, genetic counseling is advised for families to better understand the condition, recurrence risks, and available resources.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Angelman Syndrome. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about Angelman Syndrome, donation opportunities, or the progress being made on potential treatments, visit the Angelman Syndrome Foundation! The Angelman Syndrome Foundation strives “to advance the awareness and treatment of Angelman syndrome through education and information, research, and support for individuals with Angelman syndrome, their families and other concerned parties.”
Let’s keep spreading awareness! – Lily
References:
Williams, C. (2018, February 14). Angelman Syndrome – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/angelman-syndrome/

Leave a comment