What is VHL?:
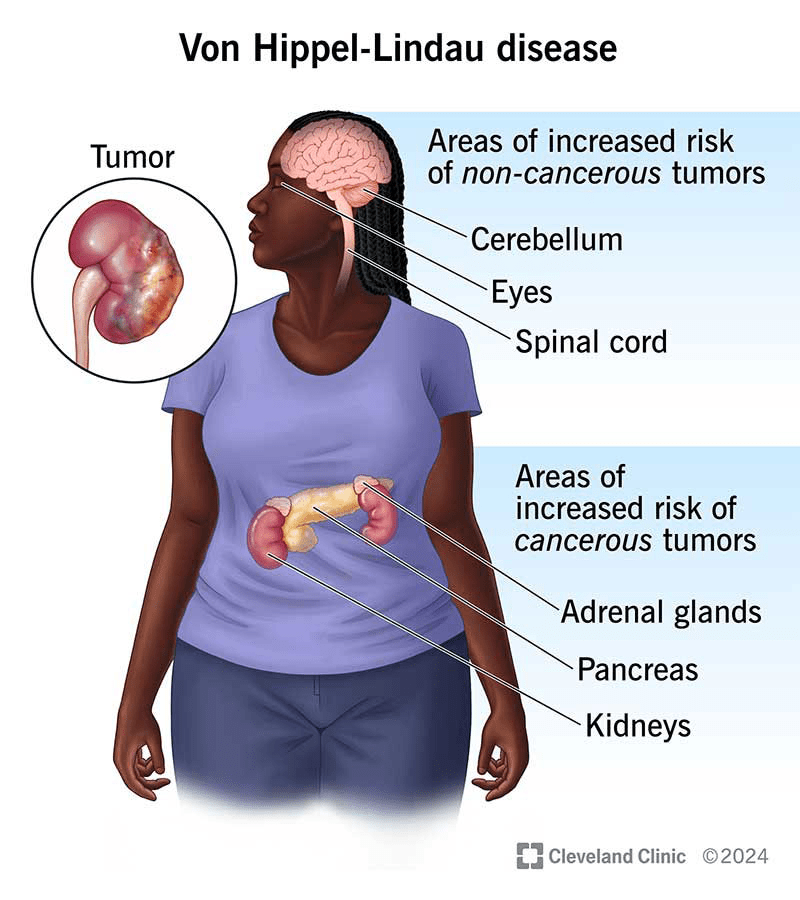
Von Hippel-Lindau (VHL) disease is a rare inherited condition that increases the risk of developing tumors in various parts of the body, such as the brain, spine, eyes, kidneys, and several other organs. While most of these tumors are non-cancerous, they can still lead to serious health issues by pressing on nearby tissues and causing symptoms like severe pain.
The disorder is caused by a mutation or deletion in the VHL gene. Its effects vary widely, even among members of the same family, making it unpredictable. Because of this, ongoing monitoring is essential throughout a person’s life to detect and manage potential complications early.
Symptoms:
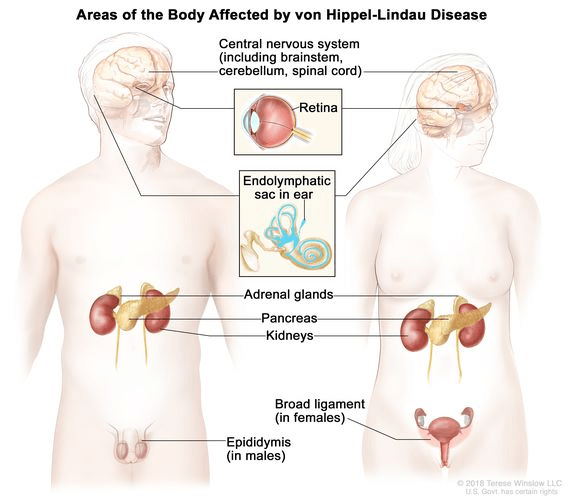
VHL does not present with a single, consistent symptom because it can affect multiple organs and occurs at different ages. It is an inherited condition, and approximately 97% of individuals with a VHL gene mutation will develop symptoms by the age of 65. However, the nature and severity of the disease can vary greatly, even among people with the same genetic variant. Some individuals may experience only mild issues, while others may develop serious complications. The average age of onset is around 26 years.
The symptoms of VHL depend on the type and location of the tumors. Hemangioblastomas, which are benign tumors commonly found in the brain, spinal cord, or eyes, can cause headaches, balance problems, muscle weakness, and vision issues such as floaters, vision loss, or retinal detachment. Adrenal gland tumors, known as pheochromocytomas, may lead to high blood pressure, panic attacks, heavy sweating, and reduced adrenal function, particularly after surgical removal. Pancreatic tumors or cysts, which can be benign or cancerous, may result in bloating, digestive discomfort, and urinary problems. Kidney tumors, particularly clear cell renal cell carcinoma, are often symptomless in the early stages but may cause lower back pain, blood in the urine, fatigue, and eventually reduced kidney function. Tumors larger than 3 cm are more likely to spread and typically require surgical removal.
Inner ear tumors, called endolymphatic sac tumors, are non-cancerous but can cause hearing loss, tinnitus, and balance issues if not treated. Less commonly, individuals with VHL may develop benign tumors in the reproductive organs, such as cystadenomas, which can impact fertility in both men and women. Tumors in the liver and lungs are generally asymptomatic and do not usually lead to significant health issues. Given the wide range of possible symptoms and organ involvement, regular screening and monitoring are essential for individuals with VHL.
Causes:
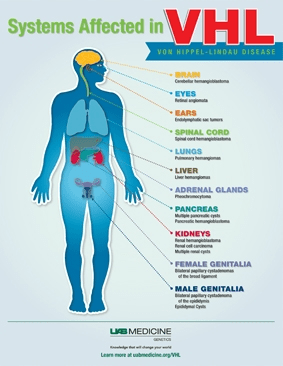
Von Hippel-Lindau (VHL) disease is a genetic disorder most commonly caused by either a deletion or a disease-causing mutation in the VHL gene, which functions as a tumor suppressor. This gene produces the VHL protein, a key component of the VCB-CUL2 complex that helps regulate normal cellular function by tagging damaged or unneeded proteins for degradation. One of its main targets is HIF-2α, a protein that regulates responses to low oxygen by activating genes involved in cell growth, red blood cell production, and blood vessel formation. When oxygen levels are normal, the VHL protein breaks down HIF-2α to prevent excessive activity of these genes. In addition to controlling HIF-2α, the VHL protein plays an important role in regulating cell division and maintaining the extracellular matrix, which supports tissue structure.
Tumor formation in VHL disease follows a “two-hit” model, where individuals inherit one mutated copy of the VHL gene, and the second mutation occurs later in a specific tissue, inactivating the remaining functional copy and allowing abnormal cell growth. VHL mutations are highly penetrant, meaning nearly all individuals with the mutation will develop related tumors by age 65. These mutations can take various forms, including missense, nonsense, frameshift, splice-site mutations, insertions, deletions, and large gene deletions. They are generally classified as truncating (which stop the protein from being fully made) or non-truncating (which alter the protein’s function without shortening it).
VHL is inherited in an autosomal dominant pattern, meaning only one copy of the altered gene is needed to cause the disease. Each child of an affected parent has a 50% chance of inheriting the mutation, and about 20% of VHL cases result from a new (de novo) mutation not inherited from either parent. In some cases, only a portion of the individual’s cells carry the mutation, a condition called mosaicism, which can affect how severe the disease is and whether it can be passed to children. VHL is classified into different types based on the types of tumors individuals develop. Type 1 usually involves large gene deletions and no pheochromocytomas (PHEOs). Type 2 involves PHEOs and is often caused by missense mutations, and is further divided into types 2A (PHEOs with low kidney cancer risk), 2B (PHEOs with kidney cancer), and 2C (PHEOs only, with no kidney cancer or brain/spinal cord hemangioblastomas). Because tumor risks vary even among individuals with the same mutation, regular screening for all types of VHL-related tumors is recommended for everyone with the condition.
Diagnosis:

Von Hippel-Lindau (VHL) disease is identified through genetic testing that detects harmful changes in the VHL gene. In some cases, a diagnosis can also be made based on the presence of characteristic tumors.
After diagnosis, early and regular monitoring is crucial because VHL-related tumors are typically more manageable when detected early. Many complications from VHL do not cause noticeable symptoms until they become serious, so proactive surveillance is key to effective treatment.
Treatment:

There is no standardized treatment plan for Von Hippel-Lindau (VHL) disease; instead, therapy is tailored to each patient based on symptoms, imaging results, lab tests, and overall health. Management strategies vary depending on the type and location of tumors.
For brain and spinal hemangioblastomas, symptoms are influenced by the tumor’s size, location, and the presence of cysts or swelling. Cysts often cause more symptoms than the tumors themselves but usually collapse once the tumor is removed. If any part of the tumor is left behind, the cyst can refill. In select cases, small, asymptomatic hemangioblastomas without cysts have been treated with stereotactic radiosurgery, although this approach is more preventive than curative and provides only limited long-term benefit. Symptoms may persist during the recovery period.
When evaluating pancreatic tumors, it is essential to distinguish between benign cysts such as serous cystadenomas and potentially harmful pancreatic neuroendocrine tumors (NETs). Cysts often do not require treatment, while NETs are assessed based on their size, behavior, and the patient’s specific VHL gene mutation.
VHL-related kidney (renal cell) carcinomas are typically found at early stages. Management focuses on careful monitoring and surgical removal only when tumors grow to about 3 cm or show rapid growth, indicating a risk of spreading. Kidney-sparing surgery is commonly used to preserve long-term kidney function. Alternatives such as radiofrequency ablation or cryotherapy may be considered for smaller tumors but require careful technique to avoid damaging nearby structures and causing scarring that may affect future surgeries.
Retinal hemangioblastomas can often be effectively treated with laser therapy when small and located at the edges of the retina. Larger lesions may require cryotherapy. Tumors on the optic disc are more difficult to treat, and preserving vision in these cases is challenging.
Pheochromocytomas, tumors of the adrenal glands, are usually removed surgically after proper preoperative medication. Even those not causing symptoms may be considered for removal, especially before pregnancy or non-emergency surgeries. Laparoscopic partial adrenalectomy is typically preferred to minimize impact on the adrenal glands.
Endolymphatic sac tumors (ELSTs) may require surgery in patients who still have hearing and show signs of tumor or bleeding on MRI. In patients who are already deaf, surgery is still recommended if other neurological symptoms are present to prevent further complications, especially balance issues. Some ELSTs are not visible on imaging and may only be detected during surgery. Cochlear implants may be considered in cases involving tumors in both ears.
In 2021, the FDA approved belzutifan (Welireg), the first drug treatment for certain VHL-associated tumors, including kidney cancer, central nervous system hemangioblastomas, and pancreatic NETs that do not require immediate surgery. Despite this advancement, surgery remains the primary treatment for most VHL manifestations. An organ-preserving approach is emphasized to avoid unnecessary tissue damage and reduce the risk of organ loss. Active Surveillance Guidelines have been developed to support early detection and timely intervention. With proper monitoring and treatment, many of the most serious effects of VHL can be significantly reduced or, in some cases, completely avoided.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Von Hippel-Lindau Disease. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about VHL, donation opportunities, or the progress being made on potential treatments, visit the VHL Alliance. The VHL Alliance strives to “[improve] quality of life and health outcomes for VHL patients, families, and caregivers with inclusive community building, connections to excellent education and treatment options, and advancements in medical research.”
Let’s keep spreading awareness! – Lily
References:
Alyea, G. (2025, May 15). Von Hippel-Lindau Disease – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/von-hippel-lindau-disease/

Leave a comment