What is GSDI?:

As a whole, glycogen storage disorders are a group of disorders affecting the metabolism of glycogen into glucose. This affects the supply of energy, as well as a person’s blood glucose level. This specific type of glycogen storage disorder is inherited in an autosomal recessive fashion. GSDI is associated with glycogen and fat buildup in the liver and kidneys, which can lead to them becoming enlarged. Short stature is also a common symptom. The mutations that cause GSDI cause enzyme deficiencies, blocking the breakdown of glycogen in specific organs. This increases the amount of glycogen and fat buildup, effectively decreasing the amount of glucose circulating through one’s bloodstream. This can also result in the accumulation of metabolites, like lactates.
Symptoms:
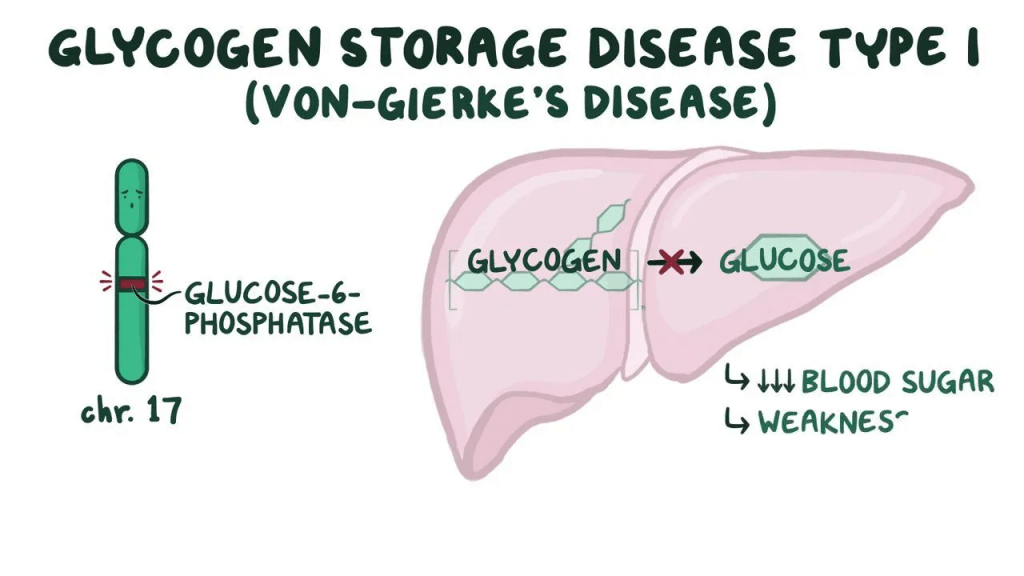
In infants with Glycogen Storage Disease Type I, low blood sugar is common. However, most symptoms begin between three to four months of age, as it takes some time for the build up to cause symptoms. The main issue in GSDI is the inability to properly manage blood sugar, leading to frequent episodes of very low blood sugar (hypoglycemia). This can cause seizures, delayed growth and development, and muscle weakness. Affected infants often have a distinctive appearance, including a “doll-like” face with chubby cheeks, a large, protruding belly due to an enlarged liver and kidneys, short stature, and thin arms and legs. In addition to low blood sugar, children with GSDI often have high levels of fats in their blood, which can lead to fatty skin bumps called xanthomas, as well as elevated levels of uric acid, increasing the risk of gout (a type of arthritis).
If left untreated, GSDI can also cause other health problems such as osteoporosis (weakened bones), delayed puberty, kidney disease, benign liver tumors, polycystic ovaries in females, inflammation of the pancreas (pancreatitis), diarrhea, and changes in brain function due to ongoing low blood sugar. The disease can also affect blood clotting, resulting in easy bruising and frequent nosebleeds. A subtype called GSD type Ib has the same symptoms as type Ia, but also includes problems with the immune system, making children more vulnerable to infections and causing chronic mouth and intestinal ulcers.
Despite these challenges, early diagnosis and proper management can significantly improve outcomes. With effective treatment, many children with GSDI grow and develop normally, live full and active lives into adulthood, and some women with the condition have had successful pregnancies.
Causes:

Type I glycogen storage disease (GSDI) is a rare inherited condition that affects how the body processes and stores sugar and fat. It happens when there’s a problem with one of two specific genes. About 80% of cases are caused by a missing or faulty enzyme called glucose-6-phosphatase, and this form is known as type Ia. The other 20% are caused by a problem with a related transporter enzyme, and this is called type Ib. In both cases, the body ends up storing too much sugar (in the form of glycogen) and fat in tissues, which can lead to health problems. The condition is passed down in families through a recessive pattern, which means a child must inherit one faulty gene from each parent to be affected. If both parents are carriers (each has one non-working copy), there’s a 25% chance with each pregnancy that their child will have the disease, a 50% chance the child will be a carrier like the parents, and a 25% chance the child won’t inherit the disease at all. Boys and girls are equally likely to be affected.
Diagnosis:
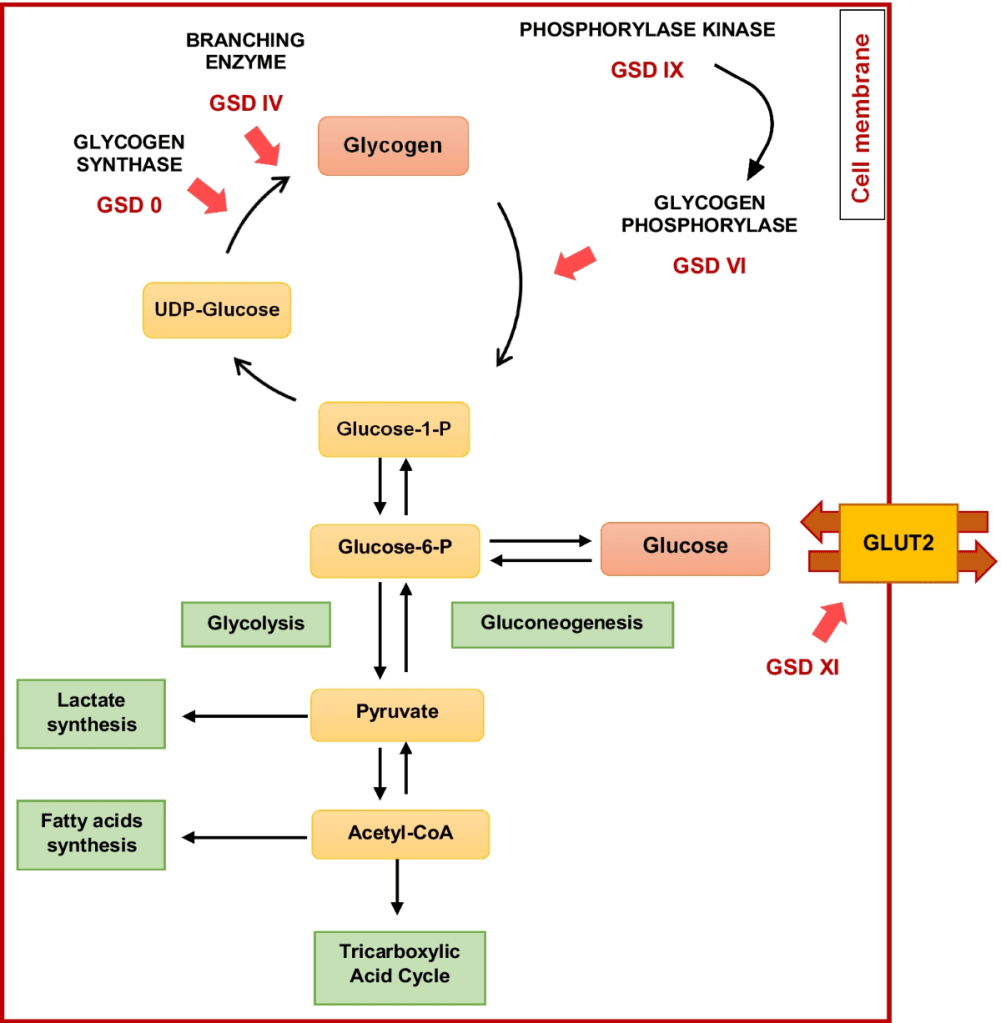
Glycogen Storage Disease (GSD) type I is diagnosed through lab tests showing abnormal levels of glucose, lactate, uric acid, triglycerides, and cholesterol. Genetic testing of the G6PC and SLC37A4 genes confirms the diagnosis and can also be used for carrier and prenatal testing. A liver biopsy may be done to confirm enzyme deficiency in GSD Ia.
Treatment:
GSD type I is managed with a specialized diet to maintain normal glucose levels, prevent hypoglycemia, and support growth. Lifelong frequent carbohydrate intake, including uncooked cornstarch, is required. Supplements like calcium, vitamin D, and iron may be needed. Medications can help manage high uric acid (e.g., allopurinol), high lipids, and kidney disease. G-CSF may treat infections in GSD Ib. Liver adenomas can be treated with surgery or radiofrequency ablation, and organ transplants may be considered if complications persist.
Patients should have annual monitoring with blood tests and kidney/liver ultrasounds. Genetic counseling is recommended for patients and their families.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop GSDI. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about GSDI, donation opportunities, or the progress being made on potential treatments, visit CureGSD. CureGSD strives to “[improve] the lives of patients and families and [advance] research towards a cure.”
References:
Bali, D., & Chen, Y.-T. (2019, December 23). Glycogen Storage Disease Type I – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/

Leave a comment