What is Mucopolysaccharides is Type 1?:
Mucopolysaccharidosis type I (MPS I) is a rare inherited disorder that affects multiple systems in the body. It results from mutations in the IDUA gene, which follows an autosomal recessive inheritance pattern, meaning both parents of an affected child are carriers. Carriers do not exhibit symptoms themselves. The disease occurs due to a deficiency or malfunction of a specific lysosomal enzyme responsible for breaking down glycosaminoglycans, which are complex carbohydrates. When these substances are not properly broken down, they build up in tissues throughout the body including the bones, joints, brain, heart, liver, and spleen. This accumulation leads to the various symptoms seen in individuals with MPS I.
MPS I is part of a broader category of conditions known as lysosomal storage disorders, which involve improper function of lysosomes. More specifically, MPS I belongs to a group of diseases known as the mucopolysaccharidoses, named for the glycosaminoglycans that accumulate in these disorders.
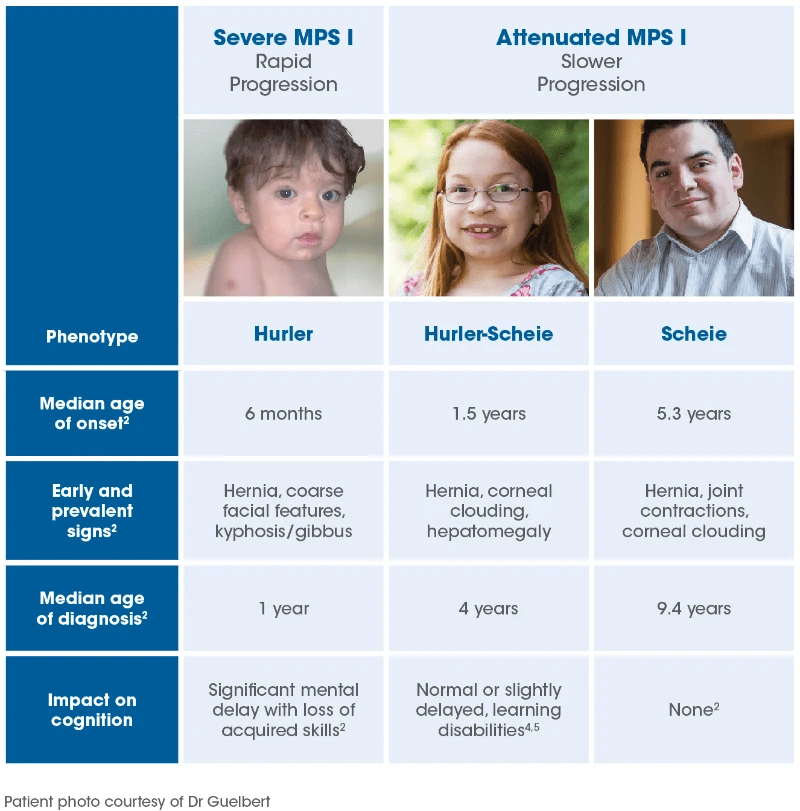
The presentation of MPS I varies widely and is best understood as a disease spectrum. Individuals are generally categorized as having either a severe or attenuated form of the condition, depending on factors such as age at symptom onset, rate of progression, and involvement of the brain. In severe cases, symptoms typically appear by six months of age and are associated with rapid progression, including early intellectual decline. Without treatment, affected individuals with the severe form often die within the first decade of life. On the other hand, the attenuated form presents later, sometimes not until after age three, and progresses more slowly. Intelligence is typically preserved in these individuals, and many can live close to a normal lifespan.
Historically, MPS I was classified into three subtypes: Hurler syndrome (severe), Hurler-Scheie syndrome (intermediate), and Scheie syndrome (mild). Today, while the term Hurler syndrome is still used for the severe form, the term attenuated MPS I is now used to encompass both the intermediate and mild presentations. This updated classification helps guide treatment options, which vary depending on the severity and progression of the disease.
Symptoms:
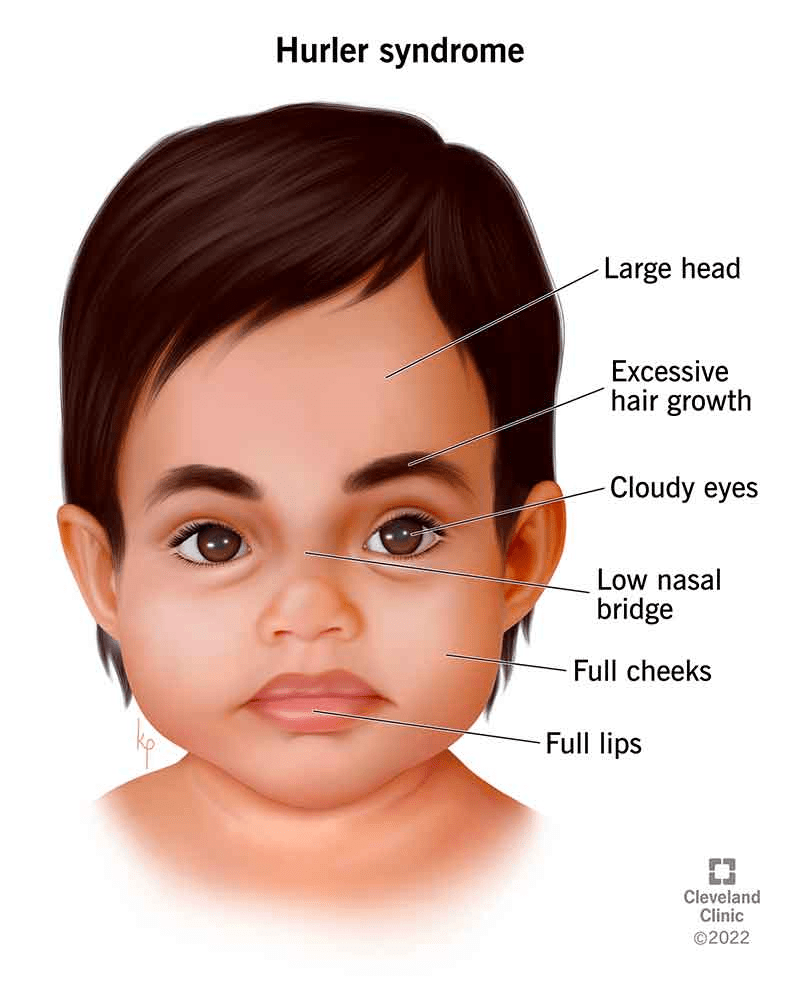
The signs and symptoms of Mucopolysaccharidosis type I (MPS I) vary widely and depend on several factors such as the specific form of the disorder (severe or attenuated), the age at which treatment begins, and the individual’s response to treatment. Since MPS I is progressive, the severity and type of symptoms often change over time and can differ significantly between individuals. Age of onset, disease progression, intellectual involvement, and physical complications are all influenced by the form of the condition.
In the severe form of MPS I, symptoms usually become noticeable between several months and one year of age. These may begin with common issues like inguinal or umbilical hernias and frequent respiratory infections, which are often overlooked as indicators of a rare disorder. As the disease progresses, affected children develop coarse facial features, including thickened lips, tongue, nasal area, and earlobes. Skull changes may result in an abnormally shaped head, and excess facial and body hair is also common. Developmental delays, particularly in speech and fine motor skills, are typical, and frequent ear infections can contribute to hearing loss.
Other early signs include stiffness in joints, skeletal deformities such as a bulging lower back, and vision issues due to corneal clouding, glaucoma, retinal degeneration, and optic nerve damage. The liver and spleen often become enlarged, though this does not usually cause symptoms on its own. As children grow, they may experience progressive intellectual decline, hydrocephalus, short stature, and worsening skeletal problems. Joint stiffness can cause claw-like hands, spinal curvature, carpal tunnel syndrome, and hip and knee deformities.
The respiratory system is often affected, with enlarged tonsils, a narrow windpipe, and sleep apnea due to airway obstruction. Heart problems are common, especially valve disease involving thickening and leaking of the mitral and aortic valves. These complications can result in blood flow issues, chest pain, shortness of breath, arrhythmias, and potentially heart failure. The heart muscle itself can also become diseased (cardiomyopathy). Eye problems, hearing loss, and ongoing physical deterioration further reduce quality of life and life expectancy if treatment is not started early.
In the attenuated form of MPS I, symptoms appear later and progress more slowly. Affected children usually do not experience early developmental delays or significant intellectual decline, which distinguishes this form from the severe type. However, many of the same symptoms can occur, including skeletal abnormalities, heart valve disease, joint stiffness, hearing loss, and eye issues like corneal clouding, glaucoma, and retinal degeneration. These may develop during childhood or adolescence and vary in severity.
Children with the attenuated form may grow more slowly and experience physical limitations due to joint and skeletal problems. Some may have mild learning difficulties, sleep apnea, hernias, and spinal cord compression, which can lead to reduced activity and fatigue. Deformities such as knock knees, high-arched feet, and toe-walking are also possible. While the rate and severity of progression vary, some individuals with the attenuated form may face serious complications by their teenage years or early adulthood. Despite a more gradual course, this form of MPS I can still have a significant impact on health and quality of life.
Causes:

Mucopolysaccharidosis type I (MPS I) is a genetic disorder caused by a mutation in the IDUA gene, which is responsible for producing the enzyme alpha-L-iduronidase. Genes serve as blueprints for making proteins, which carry out essential functions in the body. When a gene like IDUA is mutated, it can lead to the production of a protein that is either faulty, absent, or not functioning properly. In the case of MPS I, this disrupts the breakdown of complex carbohydrates known as glycosaminoglycans, particularly dermatan sulfate and heparan sulfate, which are normally broken down by alpha-L-iduronidase.
When this enzyme is deficient or absent, glycosaminoglycans accumulate inside the lysosomes of cells. This build-up interferes with normal cellular function and causes progressive tissue and organ damage. In the most severe cases of MPS I, there is a complete lack of the enzyme. In milder or attenuated forms, very small amounts of the enzyme may still be produced, which can result in a less severe presentation of the disease.
MPS I is inherited in an autosomal recessive manner, meaning that a child must inherit two altered copies of the IDUA gene, one from each parent, in order to develop the disorder. Each person has two copies of the gene, and parents of an affected child are typically carriers, having one normal and one altered copy. Carriers do not show symptoms of the disease. When both parents are carriers, there is a 25% chance with each pregnancy that the child will inherit both altered copies and be affected, a 50% chance the child will inherit one altered gene and be a carrier like the parents, and a 25% chance the child will inherit two normal genes. These probabilities are the same for both male and female children.
Diagnosis:

The diagnosis of mucopolysaccharidosis type I (MPS I) is based on the recognition of characteristic symptoms, a thorough clinical evaluation, a detailed medical and family history, and various specialized tests. It may first be suspected in infants who show early signs of the condition. In many regions, newborn screening for MPS I is currently being implemented to help with early detection.
One of the initial steps in the diagnostic process is the examination of a urine sample, which may reveal elevated levels of glycosaminoglycans such as heparan sulfate and dermatan sulfate. While this finding is not specific to MPS I, it suggests the presence of a mucopolysaccharidosis disorder. To confirm the diagnosis, more specific testing is required. This involves analyzing cells such as white blood cells or fibroblasts to measure the activity of the enzyme alpha-L-iduronidase. A significantly reduced level of this enzyme confirms the diagnosis of MPS I.
In addition to enzyme testing, molecular genetic testing is often used to identify mutations in the IDUA gene that are known to cause MPS I. This type of testing is typically conducted in specialized laboratories and serves as a confirmatory tool.
In the United States, MPS I has been added to the Recommended Universal Screening Panel for newborns, although each state decides independently when or if to implement it. One challenge of newborn screening is that it may not always be clear whether a newborn will develop a severe or attenuated form of the disorder. Since the treatment approach depends on the severity of the condition, this uncertainty can complicate early medical decisions. Ongoing research aims to improve methods for distinguishing between the severe and attenuated forms in newborns.
Treatment:
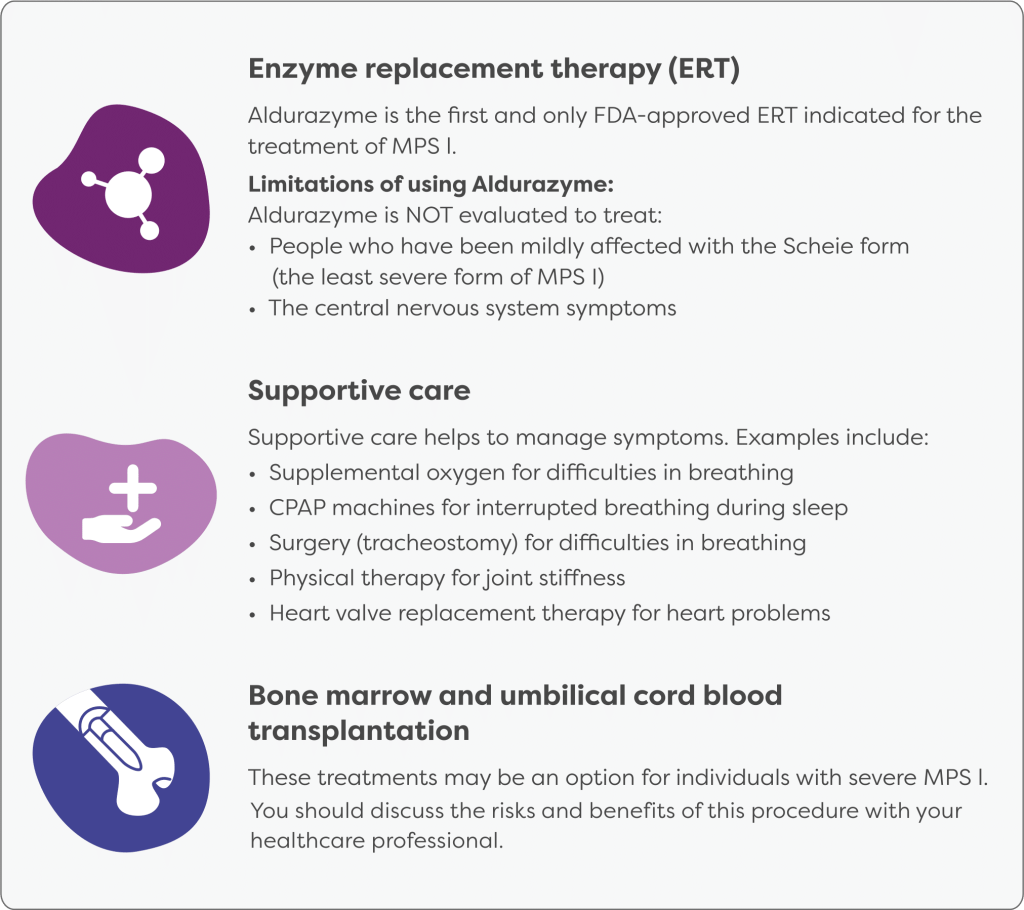
Treatment for mucopolysaccharidosis type I (MPS I) involves three main components: replacing the missing enzyme, managing symptoms, and offering genetic counseling to affected families.
The first approach is to replace the deficient enzyme, alpha-L-iduronidase. Lysosomal enzymes are unique in that they can be taken up by cells and used effectively. One method of replacement is enzyme replacement therapy (ERT), where a recombinant form of alpha-L-iduronidase is infused into the patient through a weekly intravenous procedure. In 2003, the U.S. Food and Drug Administration approved laronidase (Aldurazyme) for this purpose. Although ERT can help with many physical symptoms of MPS I, it does not affect neurological symptoms because the enzyme cannot cross the blood-brain barrier, a protective barrier that limits which substances can enter the brain.
Another method of enzyme replacement is hematopoietic stem cell transplantation (HSCT), also known as bone marrow transplantation. This involves replacing the patient’s bone marrow with healthy marrow from a donor who does not have MPS I. The transplanted stem cells produce white blood cells that make alpha-L-iduronidase, helping to restore enzyme levels in the body. HSCT is currently the standard treatment for individuals with the severe form of MPS I, as it has been shown to improve cognitive outcomes if performed early. However, the procedure is costly and carries serious risks, including potential life-threatening complications, graft-versus-host disease, and other long-term effects. While both HSCT and ERT can significantly improve the condition, neither provides a cure. Starting treatment early tends to lead to better results, and the degree of intellectual disability at the time of treatment strongly affects the overall outcome. Once intellectual impairment is established, it is usually irreversible, though further decline can be slowed.
In addition to enzyme therapy, treatment also focuses on addressing the specific symptoms each person experiences. This typically requires a coordinated team of medical specialists. Finally, genetic counseling is recommended for individuals diagnosed with MPS I and their families. This helps them understand the inheritance pattern, future family planning options, and the nature of the condition. Psychological and emotional support for the entire family is also an important part of care, helping them cope with the challenges of living with a chronic genetic disorder.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop MPS 1. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about MPS 1, donation opportunities, or the progress being made on potential treatments, visit the The Kennedy Ladd Foundation. The The Kennedy Ladd Foundation strives to “advocate and raise awareness for MPS1. To support research and fund treatments for MPS1 patients. To encourage MPS1 families to get involved and tell their stories.”
Let’s keep spreading awareness! – Lily
References:
Clarke, L. A. (2019, April 22). Mucopolysaccharidosis Type I – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/mucopolysaccharidosis-type-i/

Leave a comment