What is Alport Syndrome?:

Alport syndrome is a rare inherited condition that mainly affects the kidneys, hearing, and eyes. It causes blood in the urine early in life and gradually leads to worsening kidney function over time, which can eventually result in kidney failure. Hearing loss may develop and gradually worsen, while eye changes are common but usually do not affect vision.
There are three genetic forms of Alport syndrome. The most common type is X-linked Alport syndrome, in which males are usually more severely affected than females. Many untreated males develop kidney failure at a young age, while females often experience milder disease that appears later in life or may not progress to kidney failure at all. In the autosomal recessive form, both males and females are affected equally and often develop kidney failure during adolescence or early adulthood. The autosomal dominant form affects males and females similarly but usually progresses more slowly, with kidney problems appearing later in adulthood.
Alport syndrome is caused by mutations in genes involved in the formation of the kidney’s filtering structures. X-linked disease is caused by mutations in the COL4A5 gene, whereas autosomal forms involve mutations in the COL4A3 or COL4A4 genes. Treatment focuses on managing symptoms and slowing kidney damage with medication. Many individuals eventually require a kidney transplant.
Symptoms:
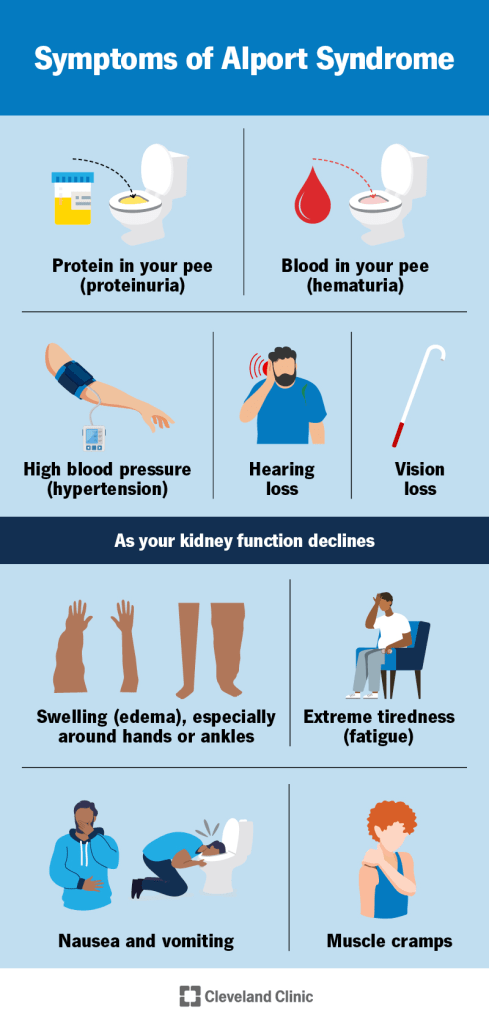
Alport syndrome varies widely from person to person, depending on the genetic type and specific gene change involved. Some individuals have a mild form that progresses slowly, while others develop serious complications earlier in life. The most common early sign is blood in the urine, which is usually only detectable under a microscope or by urine testing. Occasionally, visible blood may appear during illnesses such as colds or the flu. Blood in the urine is seen in all genetic forms of Alport syndrome and often begins in childhood.
As the condition progresses, protein begins to leak into the urine, showing that kidney damage is worsening. Over time, kidney function gradually declines and may be accompanied by high blood pressure. Eventually, some individuals develop kidney failure, which can cause fatigue, poor appetite, swelling of the legs and feet, increased thirst, and frequent urination. The speed of kidney disease progression differs by genetic type. Many untreated males with X-linked Alport syndrome develop kidney failure in their teens or early adulthood, while females are less likely to do so and usually at older ages. In autosomal recessive Alport syndrome, both males and females often develop kidney failure by early adulthood. Autosomal dominant Alport syndrome usually progresses more slowly, and many affected individuals never reach kidney failure.
Hearing loss is also common in Alport syndrome and is caused by damage to the inner ear. It usually affects both ears and worsens over time. In males with X-linked disease, hearing loss often begins in late childhood and becomes more common with age, sometimes requiring hearing aids during the teenage years. Females with X-linked disease develop hearing loss less often and usually later in life. Both males and females with autosomal recessive disease typically develop hearing loss during late childhood or adolescence, while hearing loss in autosomal dominant disease tends to occur much later, often in older adulthood.
Eye changes may also occur, especially in the X-linked and autosomal recessive forms. These can include abnormal lens shape that may require glasses and sometimes leads to cataracts, as well as changes in the retina that usually do not affect vision. Some individuals develop corneal problems that can cause eye pain, light sensitivity, blurred vision, and a gritty sensation in the eyes. In rare cases, these eye changes can lead to reduced vision. Less commonly, additional complications may occur. A small number of affected males develop bulging or weakening of the aorta, the main artery leaving the heart, which can be serious if it ruptures.
Causes:
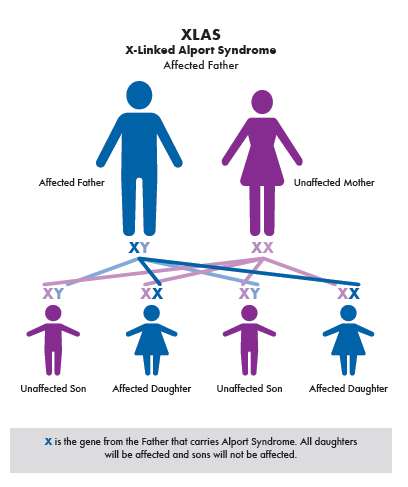
Alport syndrome is caused by harmful changes in certain genes that provide instructions for making proteins essential to normal body function. When these genes do not work properly, the proteins they produce may be missing or defective, which can damage organs such as the kidneys, ears, and eyes. The most common form is X-linked Alport syndrome, which is caused by changes in the COL4A5 gene on the X chromosome. Because males have only one X chromosome, they are more likely to develop the disease if they inherit the altered gene. Females have two X chromosomes, so those with one altered gene are often carriers and may have mild or no symptoms. Carrier females have a one in four chance with each pregnancy to have an affected son, a carrier daughter, an unaffected son, or a non carrier daughter. Affected males pass the altered gene to all of their daughters but not to their sons.
Autosomal recessive Alport syndrome occurs when a person inherits altered copies of the COL4A3 or COL4A4 gene from both parents. Parents who carry one altered gene usually have no symptoms. When both parents are carriers, each pregnancy has a one in four chance of producing an affected child, a one in two chance of producing a carrier child, and a one in four chance of producing an unaffected child. Males and females are affected equally. Autosomal dominant Alport syndrome occurs when only one altered copy of the COL4A3 or COL4A4 gene is present. The condition can be inherited from one parent or occur as a new genetic change, and each child of an affected parent has a fifty percent chance of inheriting the condition.
The severity and speed of disease progression depend on the exact gene change and its location within the gene. This relationship helps doctors estimate the risk of early kidney failure or other complications. In rare cases, larger missing sections of genetic material on the X chromosome affect multiple nearby genes, leading to Alport syndrome with additional conditions such as noncancerous smooth muscle tumors. The affected genes normally produce parts of collagen type IV, a protein that forms the structure of basement membranes in the kidneys, ears, and eyes. Basement membranes support tissues and act as filters. When collagen IV is abnormal, these membranes become weak and damaged. In the kidneys, this damage affects the filtering units, allowing blood and protein to leak into the urine. Over time, scarring develops, kidney function declines, and many individuals eventually develop kidney failure.
Diagnosis:

Alport syndrome is usually suspected based on a person’s symptoms, medical history, and a detailed clinical evaluation. The diagnosis is more likely when there is a family history of the condition, unexplained kidney failure, early hearing loss, or blood in the urine. Several specialized tests are used to confirm the diagnosis. Testing methods have changed over time, with genetic testing now playing a major role. When symptoms and family history strongly suggest Alport syndrome, genetic testing can confirm the diagnosis, identify the type of inheritance, and help predict how the disease may progress. These tests are widely available, though insurance coverage can vary. If genetic testing is not available, tissue samples may be examined instead.
A skin biopsy can help diagnose the X-linked form of Alport syndrome. This test looks for a specific collagen protein that is normally present in the skin but is often missing in affected males. Skin biopsies cannot diagnose the autosomal forms of the disease. A kidney biopsy may also be performed and can show typical changes in kidney tissue, including damage to the kidney’s filtering membrane. Special staining techniques can be used to check for missing collagen proteins in kidney tissue. Urine testing is an important part of evaluation and can detect blood in the urine, which may be constant or intermittent. As kidney disease progresses, increased levels of protein may also appear in the urine. People diagnosed with Alport syndrome should also have hearing tests and a full eye examination to check for related complications.
Treatment:

Treatment for Alport syndrome focuses on managing the symptoms each person experiences and slowing the progression of kidney disease. Care often involves a team of specialists, including kidney doctors, hearing specialists, eye doctors, pediatricians, and other healthcare providers. Genetic counseling and emotional support are also important for affected individuals and their families. Because Alport syndrome is rare, large treatment studies have been limited, but clinical guidelines based on expert experience are available. Medications called ACE inhibitors are commonly used and have been shown to slow kidney damage and delay kidney failure by reducing protein loss in the urine and lowering blood pressure. These medications are usually recommended once protein is clearly present in the urine and may be considered earlier in some cases. Not everyone can tolerate ACE inhibitors, so alternative medications called angiotensin receptor blockers may be used instead.
Although these treatments can slow kidney disease, there is no cure for Alport syndrome, and kidney function often continues to decline over time. When kidney failure occurs, dialysis or a kidney transplant is needed. Dialysis replaces some kidney functions by filtering waste and balancing fluids and minerals, either through a machine or using fluid in the abdomen. Kidney failure is permanent, so long-term dialysis or transplantation is required.
A kidney transplant is the preferred treatment for people with Alport syndrome who reach kidney failure and usually has very good outcomes. The age at which a transplant is needed varies widely, and many individuals, especially females with X-linked disease and some with autosomal dominant disease, may never need one. Careful donor selection is important to avoid using relatives who may also carry the condition. Alport syndrome does not return in the transplanted kidney, though a small number of patients may develop immune-related complications. Other symptoms are treated using standard medical care. Hearing loss is managed with hearing aids, which are often very effective, and cataracts are treated with surgery when necessary.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Alport syndrome. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about Alport syndrome, donation opportunities, or the progress being made on potential treatments, visit the Alport Syndrome Foundation. The Alport Syndrome Foundation strives “to improve the lives of people living with Alport syndrome through education, empowerment, advocacy, and direct investment in research.”
References:
Kashtan, C. (2024, March 25). Alport Syndrome – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/alport-syndrome/

Leave a comment