What is Apert Syndrome?:
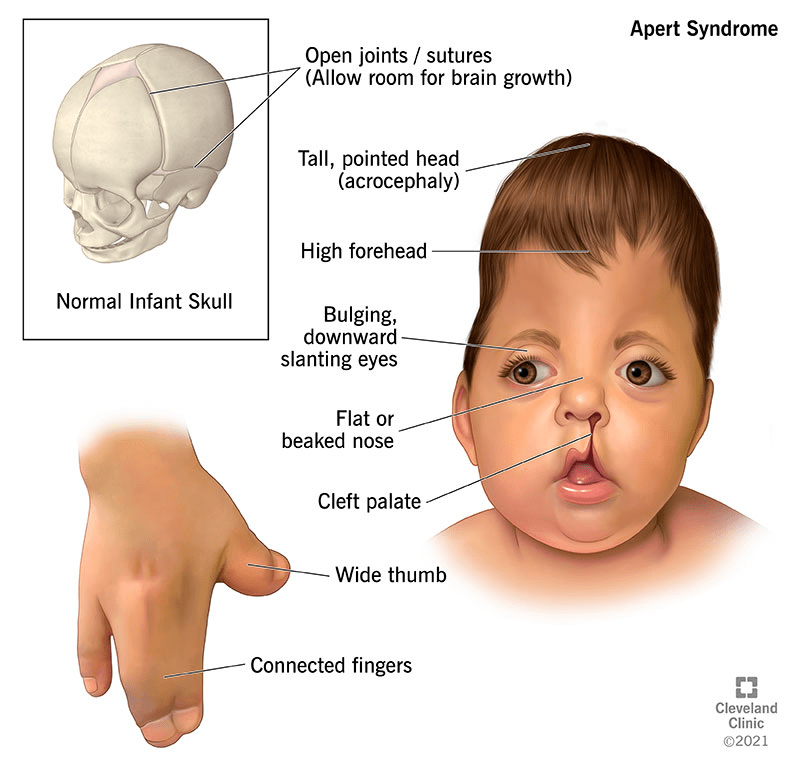
Apert Syndrome, or acrocephalosyndactyly type 1, is a rare congenital disorder characterized by premature fusion of skull sutures, causing distinctive facial and head malformations, as well as fused or webbed fingers and toes. Often featuring a pointed head, sunken midface, connected fingers, and intellectual disability, this condition typically results from random genetic mutations rather than inheritance. While symptoms and their severity vary, treatment generally involves specialized, multi-stage reconstructive surgeries for the skull, face, and limbs.
Symptoms:
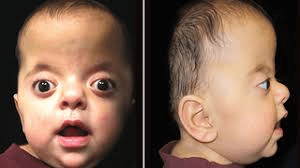
Apert syndrome is a rare genetic disorder characterized by craniosynostosis, where premature fusion of skull sutures causes a pointed head shape (acrocephaly), flattened back of the head, and increased intracranial pressure, sometimes leading to hydrocephalus. This condition causes significant craniofacial abnormalities, including widely spaced or bulging eyes, underdeveloped midface, and dental issues. Furthermore, affected individuals may face serious health risks like breathing/swallowing difficulties, sleep apnea, and potential infections due to airway obstructions.
Apert syndrome is a rare genetic disorder characterized by the premature fusion of skull bones and severe, complex syndactyly of the hands and feet, with upper limbs typically more severely affected than lower limbs. Affected individuals often present with a “mitten-like” appearance of the hands, featuring fused fingers, stiff joints, and broad thumbs, as well as fused toes. Beyond these characteristic skeletal and limb malformations, the condition can cause short stature, fusion of neck vertebrae, and various neurological issues, including intellectual disability ranging from mild to moderate and structural brain anomalies. Other potential health complications include hearing loss, chronic ear infections, cardiac septal defects, a narrowed stomach-intestinal opening, blockage of the esophagus, an out-of-position anus, blockage of the vagina, failure of the testicles to fall, and enlarged kidneys due to blockage.
Causes:

Apert syndrome is a genetic disorder caused by mutations in the FGFR2 gene, specifically Ser252Trp or Pro253Arg, which disrupts essential skeletal development, particularly the normal, timely closure of skull sutures. This mutation causes improper signaling in fibroblast growth factors, leading to characteristic malformations such as premature skull fusion, distinctive facial features, and webbed hands or feet. While related to other syndromes like Crouzon and Pfeiffer, Apert syndrome presents with distinct clinical features resulting from this specific, mostly spontaneous, genetic alteration.
Apert syndrome is a rare genetic disorder primarily caused by new, random mutations in the FGFR2 gene in up to 95% of patients, with a higher incidence observed in cases with advanced paternal age. While it can rarely be inherited in an autosomal dominant manner, where a single mutated copy causes the condition, most cases arise without family history. When inherited, there is a 50% risk of transmission from an affected parent to their offspring, with equal risk for both males and females.
Diagnosis:
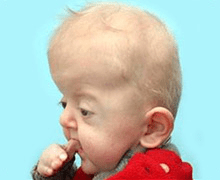
Apert syndrome is typically diagnosed at birth or during infancy through clinical evaluation of physical features like craniosynostosis, facial anomalies, and syndactyly. Confirmation often involves FGFR2 gene mutation testing, while imaging tools such as CT scans and MRIs assess skeletal and brain abnormalities. Prenatal diagnosis is also possible via 2D/3D ultrasound or fetal MRI to detect these characteristics.
Treatment:
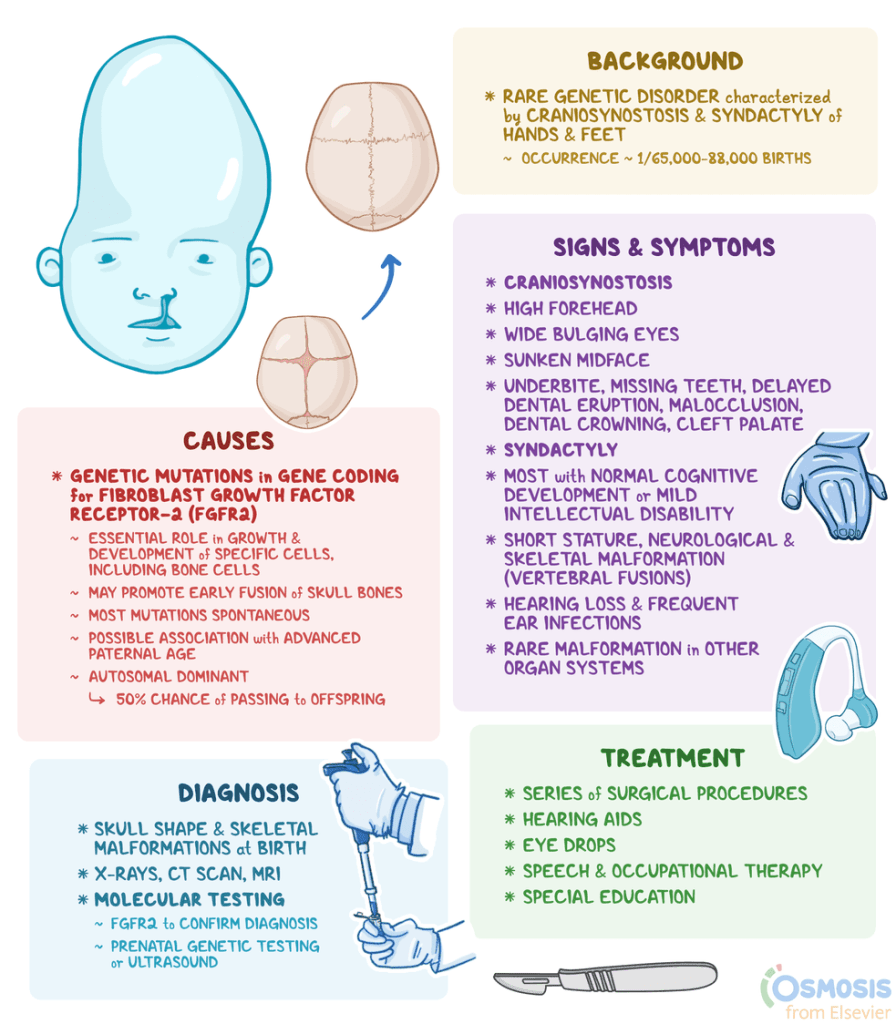
The treatment of Apert syndrome is tailored to individual symptoms and typically requires a multidisciplinary team of specialists, including pediatricians, surgeons, neurosurgeons, orthopedists, otolaryngologists, and cardiologists. Because the condition involves craniosynostosis and hydrocephalus, which can increase pressure on the brain, early surgical intervention, often within two to four months of birth, is commonly advised to correct skull abnormalities and insert shunts to drain excess cerebrospinal fluid. These symptomatic and supportive therapies aim to manage the specific, varied manifestations of the syndrome, such as bone fusion, to improve the child’s development and quality of life.
Corrective and reconstructive surgery is often recommended to address craniofacial malformations, polydactyly, syndactyly, and other skeletal or physical abnormalities associated with Apert syndrome, with treatments for cardiac or hearing issues also sometimes necessary. Early intervention, including specialized services such as physical therapy and occupational therapy, is crucial for helping children reach their full potential. Furthermore, genetic counseling is highly recommended for affected individuals and their families to understand the condition’s causes and risks for future children. At the same time, comprehensive psychosocial support is essential for the entire family.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Apert Syndrome. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disorder. If you’re interested in learning more about Apert Syndrome, donation opportunities, or the progress being made on potential treatments, visit the Foundation for Faces of Children. The Foundation for Faces of Children’s mission is to “[develop] funding for teaching materials and information for parents.”
Let’s keep spreading awareness! – Lily
References:
Cahn, S. (2019, July 30). Apert Syndrome – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/apert-syndrome/

Leave a comment