What is Rett Syndrome?:

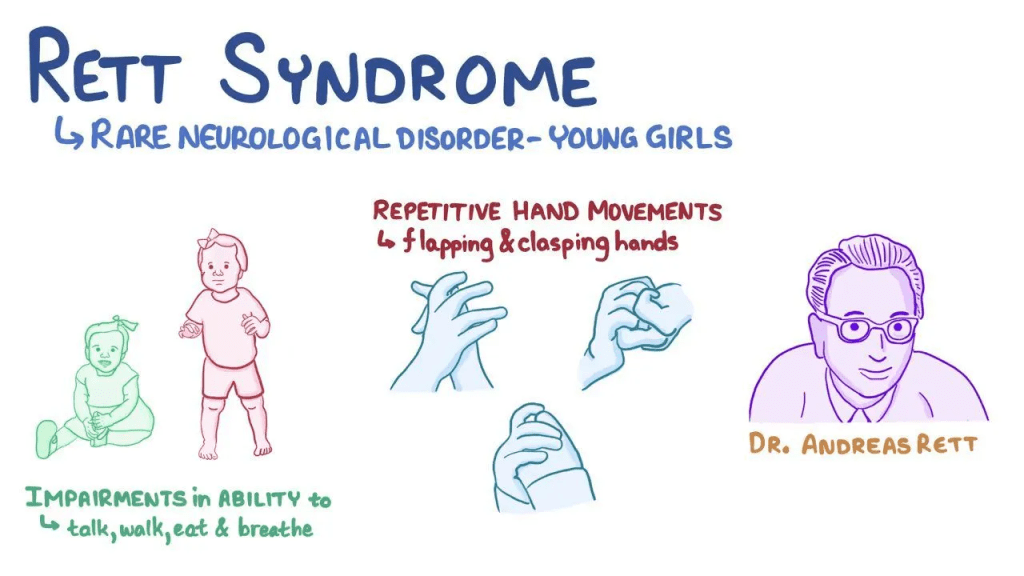
Rett syndrome is a progressive neurodevelopmental condition that primarily affects females and only rarely occurs in males. Infants with Rett syndrome usually grow and develop normally during the first 7 to 18 months of life. After this period, they begin to lose previously learned skills, including purposeful hand use and the ability to communicate. As the disorder progresses, additional problems appear, such as difficulty controlling voluntary movements and repetitive, involuntary hand actions like clapping or rubbing.
Many children with Rett syndrome also develop slowed head growth. Other common features include behaviors similar to autism, irregular breathing, feeding and swallowing problems, delayed growth, and seizures. Most cases are caused by mutations in the MECP2 gene located on the X chromosome. The severity of the disorder can vary widely, ranging from mild to severe disabilities. The course of the condition depends on the specific type and location of the MECP2 mutation and on a biological process called random X inactivation. Because of this, two girls of the same age with the same mutation may show very different symptoms and levels of impairment.
Rett syndrome was first described in the 1960s by Austrian physician Andreas Rett. Researchers now view it as part of a broader group of conditions related to MECP2 gene mutations, known as MECP2-related disorders. This group includes classic Rett syndrome, variant Rett syndrome, MECP2-related severe neonatal encephalopathy, and PPMX syndrome. Another related condition is MECP2 duplication syndrome, which results from an extra copy of the MECP2 gene on the X chromosome.
Symptoms:
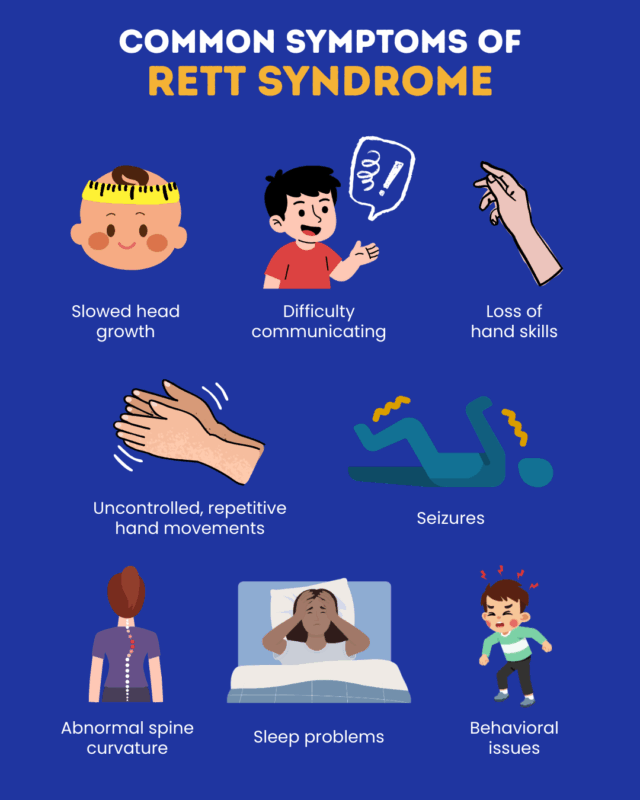
The symptoms, progression, and severity of Rett syndrome vary greatly from person to person. The disorder mainly affects females and is considered part of a spectrum of conditions linked to mutations in the MECP2 gene. Individuals may experience different combinations of symptoms, and not everyone develops every feature of the disorder. Symptoms usually appear gradually in stages. Most infants with classic Rett syndrome appear to develop normally during the first 6 to 18 months of life. However, subtle signs may appear earlier. Some infants are unusually calm, have weak crying, poor sucking ability, or low muscle tone. Head growth may begin slowing as early as three months, which can lead to acquired microcephaly, meaning the head is smaller than expected.
Between about 6 and 18 months, development may slow or stop. Infants may lose eye contact, show less interest in toys or play, and appear less responsive to people. Irritability, crying, and restlessness may occur. Some developmental progress may still happen, but at a slower rate. For example, a child might learn to sit but not crawl. Between about 1 and 4 years of age, children begin losing previously acquired skills, especially spoken language and purposeful hand movements. Social interaction may decline, and intellectual functioning may worsen. This change can happen quickly or gradually.
During this stage, a defining feature of Rett syndrome appears: repetitive, involuntary hand movements such as wringing, squeezing, clapping, rubbing, or bringing the hands to the mouth. Episodes of intense crying or screaming may also occur. Other symptoms may develop, including autistic-like behaviors, panic attacks, teeth grinding, tremors, and apraxia, which is difficulty performing learned movements even when the person understands the command. Apraxia can also affect communication. Seizures are common, and some children develop balance problems due to poor coordination when walking.
Breathing abnormalities while awake may also appear, including unusually slow or rapid breathing, breath holding, swallowing air, or brief pauses in breathing. Stress can make these breathing problems worse. After the period of rapid regression, neurological symptoms often stabilize. Some individuals may show mild improvements in eye contact, behavior, communication, and social interaction. However, many symptoms continue, including repetitive hand movements, seizures, teeth grinding, and breathing irregularities. Intellectual ability is difficult to assess because many individuals cannot speak or use their hands effectively.
After about age 10, many individuals experience worsening movement difficulties. Some may never learn to walk, while others may lose the ability later in life. Muscle weakness, joint stiffness, and spasticity can develop, causing slow and stiff movements. Hands and feet may remain small, underdeveloped, and often cold. Many individuals develop dystonia, which causes sustained muscle contractions and abnormal movements. Some may also show symptoms similar to Parkinson’s disease, such as reduced facial expression, stiffness, and tremors.
Around 85 to 90 percent of individuals experience growth problems and muscle wasting, often caused by difficulties chewing and swallowing that limit food intake. In some cases, especially when more function is retained, individuals may overeat and become obese. Other possible complications include digestive problems such as constipation, bowel movement difficulties, acid reflux, and enlargement of the colon. Additional issues can include cold hands and feet, crossed eyes, scoliosis, gallbladder problems, gallstones, and reduced bone density, which can make bones fragile.
Many people with Rett syndrome live into adulthood but usually require ongoing care and supervision. However, there is a higher risk of sudden death compared with the general population. About one quarter of deaths occur suddenly and unexpectedly, sometimes due to heart rhythm abnormalities. These can involve a prolonged QT interval, which affects the electrical activity that controls the heartbeat and may lead to irregular heart rhythms.
Causes:
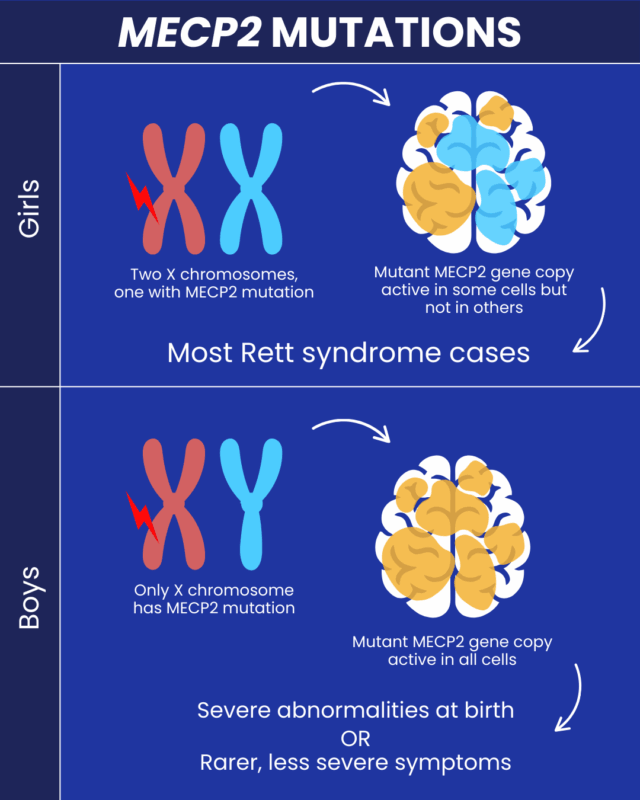
About 90 to 95 percent of Rett syndrome cases are caused by mutations in the MECP2 gene, and more than 200 different mutations of this gene have been identified. In roughly 99 percent of cases, these mutations occur spontaneously rather than being inherited from a parent. This means that most parents of a child with Rett syndrome have normal chromosomes, and the mutation develops in one of the parents’ reproductive cells, most often on the father’s side.
For parents who already have one child with Rett syndrome, the chance of having another affected child is about 1 percent. In rare situations, more than one child in a family may be affected due to germline mosaicism. This occurs when some of a parent’s reproductive cells carry the MECP2 mutation while the rest of the body’s cells do not. Because the mutation is only present in certain reproductive cells, the parent shows no symptoms but can still pass the mutation to one or more children. The likelihood of passing on the mutation depends on how many of the parents’ germ cells carry it. There is currently no test to detect germline mosaicism before pregnancy, although prenatal testing may be available through genetic counseling.
In very rare cases, Rett syndrome may be inherited from a mother who carries the MECP2 mutation but has no symptoms or only mild ones. This can happen when the normal X chromosome is more active than the mutated one due to a process called random X chromosome inactivation. If a mother is a known carrier of the mutation, each child has a 50 percent chance of inheriting it. Random X chromosome inactivation is a natural process in females, who have two X chromosomes, while males have one X and one Y chromosome. In each female cell, one X chromosome is randomly turned off. Sometimes the chromosome carrying the mutation is mostly silenced, which can lead to mild symptoms. In other cases, the healthy X chromosome may be mostly silenced, resulting in more severe symptoms.
Because males only have one X chromosome, mutations in the MECP2 gene are usually fully expressed in males. In many cases, this mutation is not compatible with life and may lead to miscarriage or stillbirth. Some females with an MECP2 mutation may not show symptoms, suggesting that other biological factors, such as modifier genes, may reduce the effects of the mutation. Researchers continue to study these mechanisms to gain a deeper understanding of the condition.
The MECP2 gene is located on the long arm of the X chromosome at position Xq28. Chromosomes carry genetic information in the nucleus of human cells, and most human cells contain 46 chromosomes arranged in pairs. The MECP2 gene provides instructions for producing a protein called methyl-CpG binding protein 2, which helps regulate the activity of many other genes. When the MECP2 gene is mutated, the body produces too little functional MECP2 protein. This disrupts the normal regulation of other genes, allowing some genes that should be inactive to remain active during development. These changes are believed to interfere with normal brain development in early childhood, although the exact mechanisms are still not fully understood.
Diagnosis:
Rett syndrome is diagnosed by identifying its characteristic symptoms, reviewing a detailed medical history, and performing a thorough clinical evaluation. Doctors may also conduct specialized tests to rule out other conditions that could cause similar symptoms. Genetic testing can be used to detect mutations in the MECP2 gene, which can confirm the diagnosis.
Treatment:
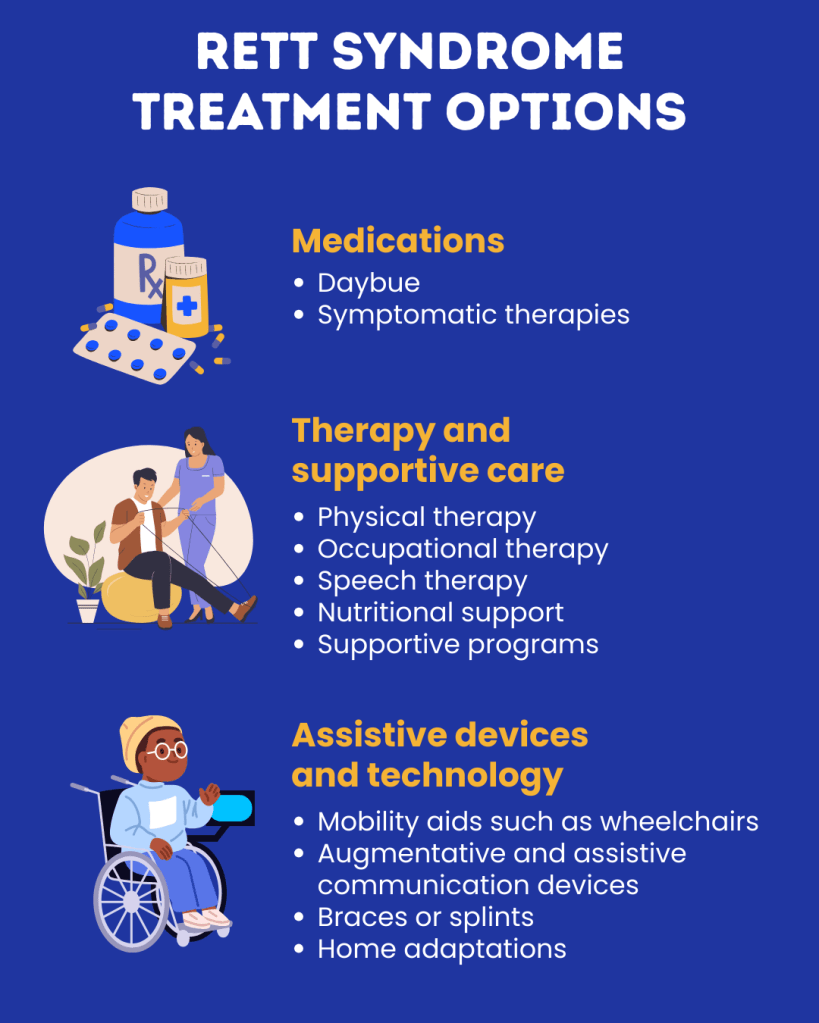
Treatment for Rett syndrome focuses on managing the specific symptoms each individual experiences. Care usually requires a multidisciplinary team of healthcare professionals, which may include pediatricians, neurologists, gastroenterologists, speech therapists, psychiatrists, nutritionists, and other specialists. Genetic counseling can also be helpful for affected individuals and their families.
Treatment plans are highly individualized and often involve multiple therapies. Early developmental intervention is important to help children reach their full potential. Many children benefit from occupational therapy, physical therapy, and speech therapy, along with behavioral and rehabilitative therapies. Additional services such as special education, social support, or vocational assistance may also be needed. Emotional and psychosocial support for the family is also an important part of care. Most other treatments are supportive and aimed at controlling symptoms. For example, nutritional support may be necessary to ensure adequate calorie intake, and some children may require a feeding tube placed in the stomach. High-calorie and high-fat diets are sometimes recommended.
Medications may be prescribed to treat symptoms such as seizures, anxiety, sleep problems, breathing irregularities, repetitive hand movements, gastrointestinal issues, muscle stiffness, and spasticity. Because some individuals with Rett syndrome are at risk for heart rhythm problems such as a prolonged QT interval, evaluation by a cardiologist may be recommended, and certain medications that worsen this condition should be avoided. Scoliosis is also common in Rett syndrome, and specific clinical guidelines have been developed to help manage spinal curvature in affected individuals.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Rett Syndrome. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about Rett Syndrome, donation opportunities, or the progress being made on potential treatments, visit the International Rett Syndrome Foundation. The International Rett Syndrome Foundation’s mission is to “accelerate full spectrum research to cure Rett syndrome and empower families with information, knowledge, and connectivity.”
References:
Neul, J. L., & Eskind, A. S. (2023, March 15). Rett Syndrome – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/rett-syndrome/

Leave a comment