What is FFI?:
Fatal familial insomnia (FFI) is a rare, fatal neurodegenerative prion disease caused by a misfolded PRNP gene, which creates toxic protein build-up, primarily in the thalamus. Characterized by severe insomnia, the disorder causes rapid physical and mental deterioration, including autonomic nervous system failure, hallucinations, and dementia. FFI currently has no cure.
Symptoms:
FFI is characterized primarily by a sudden, middle-aged onset of rapidly worsening insomnia that results in severe sleep deprivation and eventual coma or death. While inability to sleep is the hallmark, patients may also present with progressive dementia, exhibiting significant cognitive declines in memory, attention, and speech. Early signs often include weight loss and inattentiveness, later progressing to hallucinations, confusion, and potentially ataxia.
Common symptoms include: diplopia, nystagmus, dysphagia, dysarthria, ataxia, myoclonus, and other symptoms similar to those experienced by people with Parkinson’s disease. Other symptoms vary from person to person but may include: fever, tachycardia, hypertension, hyperhidrosis, increased tear production, constipation, changes in body temperature, sexual dysfunction, anxiety, and depression.
Causes:
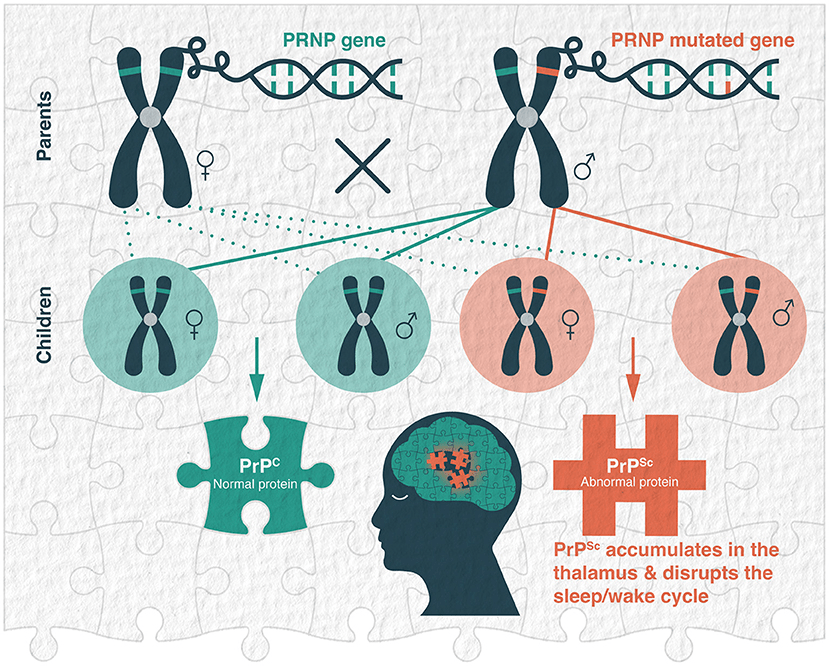
FFI is an autosomal dominant disease caused by a mutation of the PRNP gene located on chromosome 20. This mutation leads to the production of faulty prion proteins (PrPSc) that misfold, cluster in the thalamus, and become toxic to the central nervous system, resulting in severe sleep disruption and neurodegeneration. While FFI is typically inherited from an affected parent, it can, in rare instances, occur spontaneously as a de novo mutation in an individual without a family history. Someone with a new de novo variant can pass the mutated gene to their children in an autosomal dominant manner. Dominant genetic disorders require only one mutated gene from either parent, carrying a 50% risk of inheritance for each child, regardless of sex. Conversely, Sporadic Fatal Insomnia (SFI) is a rare and random occurrence of fatal insomnia that happens without the PRNP gene mutation, distinguishing it from familial forms.
The resulting progressive destruction of this brain region caused by the buildup of PrP causes severe, fatal neurodegeneration, often appearing as sponge-like gaps. Coined to represent a “proteinaceous infectious agent,” a prion is an abnormally misfolded version of the protein PrP (PrPSc) that acts as the transmissible agent of fatal neurodegenerative prion diseases, such as Kuru and variant Creutzfeldt-Jakob disease. While these conditions can be “acquired” through direct exposure to or ingestion of diseased brain tissue, they are not contagious in the traditional sense. Furthermore, while some cases are familial, the majority of prion diseases are classified as sporadic.
Diagnosis:
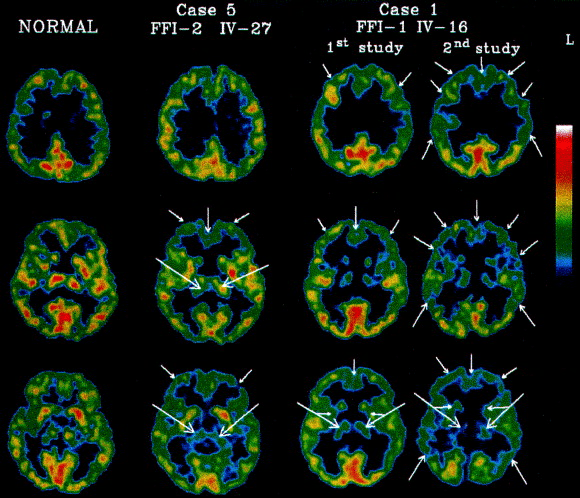
A diagnosis of FFI is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation, and a variety of specialized tests. Molecular genetic testing available at specialized laboratories can confirm a diagnosis by detecting an abnormal variant in the PRNP gene, which is present in all FFI cases. While this genetic testing can confirm the disorder, a negative result does not rule out SFI, making a detailed evaluation essential. Additionally, polysomnography is commonly used to demonstrate significantly reduced sleep duration and difficulties in transitioning through different sleep stages.
Positron emission tomography (PET) scan is an advanced, three-dimensional imaging technique that measures brain metabolism to aid in diagnosing FFI or SFI, often highlighting characteristic reduced activity in the thalamus. While MRI may show supportive, yet not fully characterized, signs for prion diseases and CT scans generally lack diagnostic utility for FFI, both MRI and CT remain useful for ruling out mimicking conditions.
Tests for the 14-3-3 protein in cerebrospinal fluid (CSF) are used to detect rapid neuronal death, often indicating FFI or other prion diseases, though normal levels do not rule out FFI. While 14-3-3 is a key biomarker, elevated CSF tau protein may also be present. Additionally, the RT-QuIC test is increasingly used to detect low levels of prions, providing a more specific diagnostic tool, though data on its efficacy for FFI is still emerging.
Treatment:
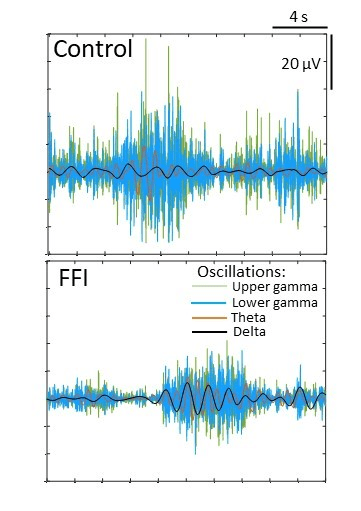
FFI is an incurable disease requiring a coordinated team of specialists to manage symptoms and provide palliative care. Due to the rarity of the disease, there are no standardized treatment protocols, and management is tailored to the individual, often involving anticonvulsants for seizures or twitching, and the discontinuation of medications that worsen confusion or insomnia. While sleep aids are often ineffective long-term, comprehensive care, including psychosocial support and genetic counseling, is crucial for both patients and their families. While research is ongoing to find effective therapies to slow or stop disease progression, currently, no effective treatment exists to stop FFI from being fatal.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Fatal Familial Insomnia. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about FFI, donation opportunities, or the progress being made on potential treatments, visit the Creutzfeldt-Jakob Disease Foundation. The Creutzfeldt-Jakob Disease Foundation’s mission is to “support families affected by Prion Disease, raise awareness, and support medical education and research.”
References:
Mastrianni, J. A., & McLoraine, H. (2023, July 17). Fatal familial insomnia – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/fatal-familial-insomnia/

Leave a comment