What is Moyamoya Disease?:
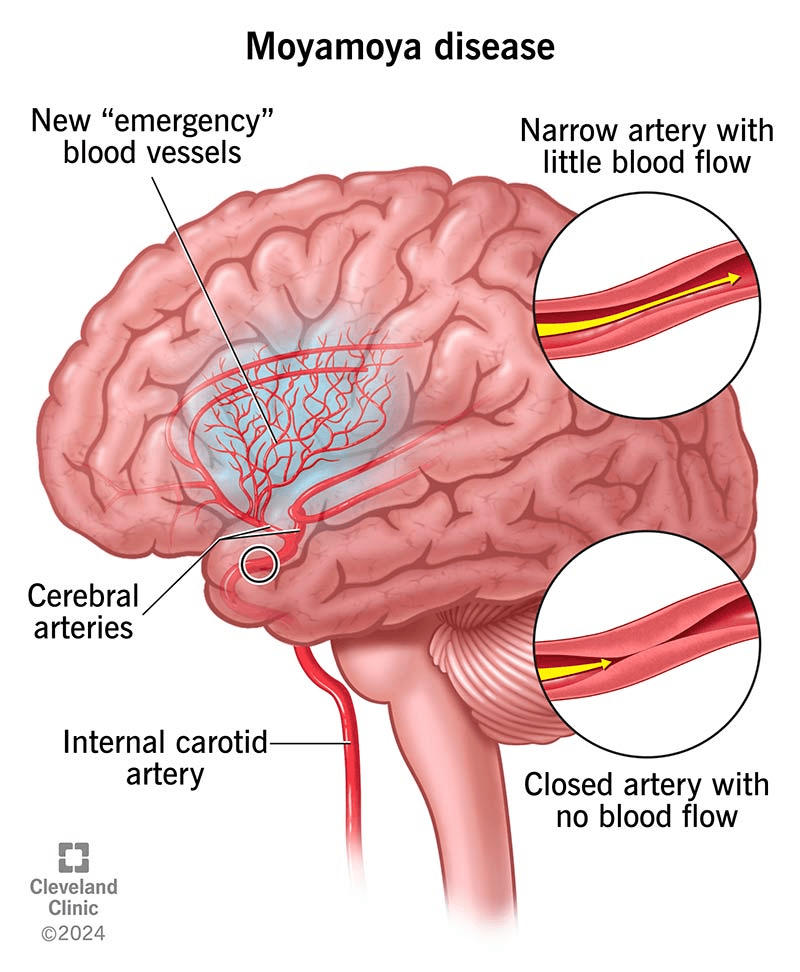
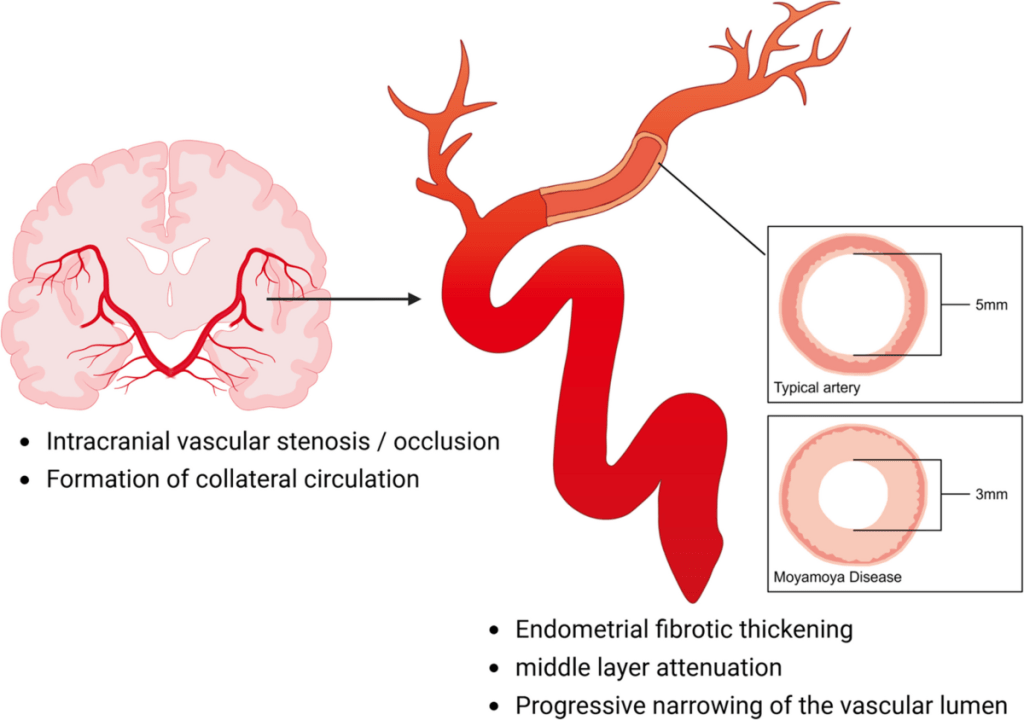
Moyamoya disease is a progressive cerebrovascular disorder defined by the narrowing or total blockage of the carotid arteries within the skull. As these primary pathways for blood flow constrict, the brain attempts to compensate by developing a cluster of fragile, tiny collateral blood vessels at its base. These compensatory vessels create a distinct “puff of smoke” appearance on imaging, which is the literal translation of the Japanese word moyamoya. Because these vessels are often insufficient, the resulting lack of oxygen to the brain can trigger serious neurological events, most notably strokes or transient ischemic attacks that cause paralysis, speech loss, or sensory impairment.
Beyond the risk of stroke, the condition can manifest through headaches, seizures, visual disturbances, and developmental delays. While the disease is considered idiopathic in many patients, there is a strong genetic component in 10% to 30% of Asian cases. Clinically, a distinction is made between “moyamoya disease,” which occurs on a familial or isolated basis, and “moyamoya syndrome,” which appears alongside underlying conditions like sickle cell disease or Down syndrome. Regardless of the classification, the primary concern remains the chronic deprivation of oxygen to brain tissue.
Symptoms:
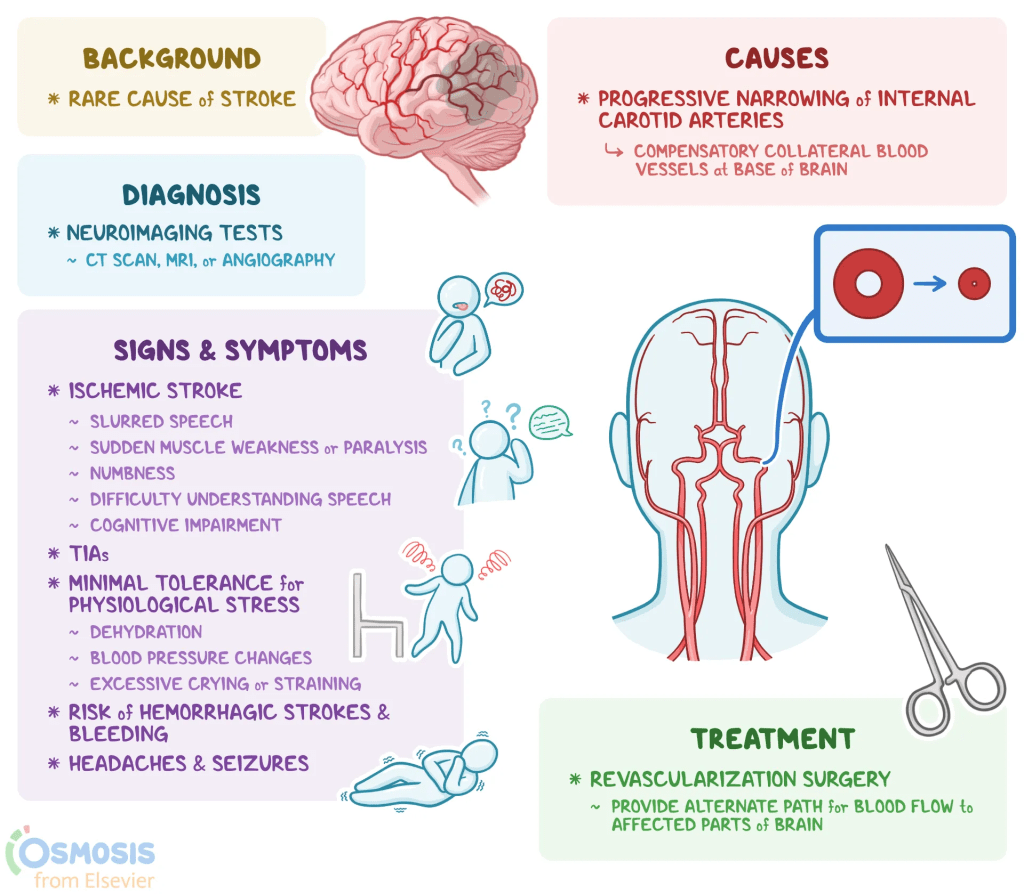
Moyamoya disease typically manifests during two distinct life stages: early childhood and mid-adulthood. In children, the condition most frequently presents through symptoms tied to reduced blood flow, such as strokes, TIAs, seizures, or developmental delays. While adults also experience these ischemic symptoms, they face a significantly higher risk of intracranial hemorrhage. This increased vulnerability to bleeding is likely caused by the rupture of fragile moyamoya vessels, often exacerbated by the higher blood pressure levels commonly found in adult patients.
Causes:
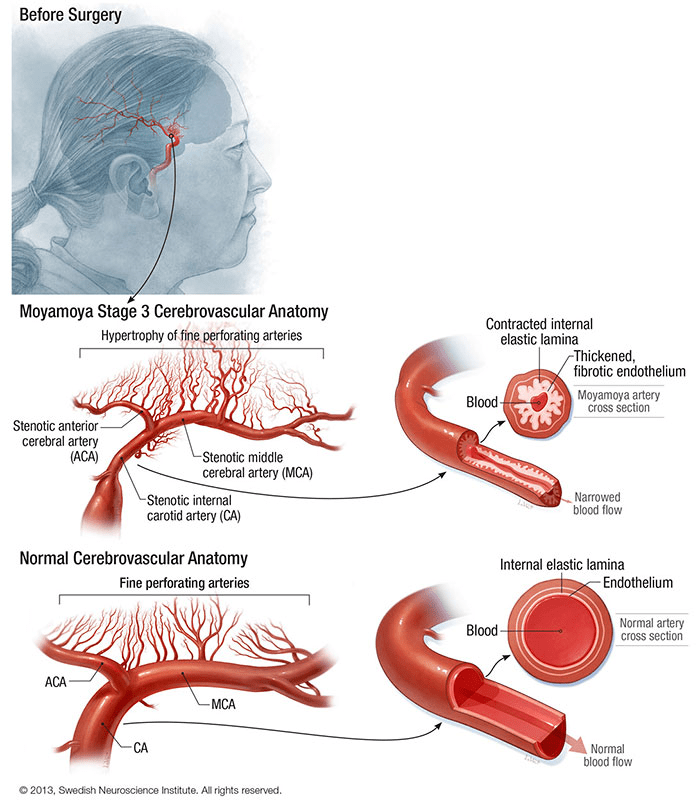
The exact causes of moyamoya disease remain partially shrouded in mystery, but research indicates a significant genetic foundation, particularly in East Asian populations. The primary driver is the RNF213 gene, specifically the R4810K variant, which is present in the vast majority of cases in Japan and South Korea. This gene is responsible for regulating angiogenesis, the growth and repair of blood vessels. While the disease is typically inherited in an autosomal dominant pattern, it exhibits incomplete penetrance, meaning many people carry the genetic variant without ever developing symptoms. Interestingly, inheritance patterns suggest epigenetic factors like genomic imprinting may be at play, as the disease is more frequently passed down from mothers to daughters.
In addition to RNF213, scientists have identified several other genes, such as ACTA2, DIAPH1, and GUCY1A3, which affect the structural integrity and relaxation response of blood vessel walls. Despite these discoveries, known genetic variants currently account for less than 2% of all cases globally, and the specific mutations found in White populations differ significantly from those in Asian populations. This suggests that while genetics provides a baseline susceptibility, the condition likely requires an environmental or external “trigger” to manifest.
The complexity of moyamoya is further highlighted by the fact that even identical twins do not always both develop the disease. This point underscores the influence of non-genetic risk factors, including radiation exposure, infections, and autoimmune disorders. Because of the low penetrance of associated genes and the high degree of variation between different ethnic groups, genetic testing is not yet a definitive diagnostic tool. Instead, the disease is viewed as a multifaceted interplay between an individual’s DNA and their environment, affecting critical biological pathways like inflammation and vascular remodeling.
Diagnosis:
The diagnosis of moyamoya disease is primarily achieved through non-invasive imaging, specifically MRI and MRA scans with a magnet strength of at least 1.5 Tesla. While the MRI evaluates the brain tissue for signs of stroke or reduced blood flow, the MRA focuses on the vasculature to identify the arterial narrowing and characteristic “puff of smoke” collateral vessels. Under current clinical guidelines, these scans are often sufficient to establish a definitive diagnosis without the need for more invasive procedures.
However, cerebral catheter angiography remains a critical diagnostic tool for complex cases and surgical preparation. While MRI and MRA confirm the disease’s presence, angiography provides a more precise map of blood flow patterns and the exact degree of vessel blockage. Most importantly, it allows surgeons to identify transdural collaterals, which are vital for determining the surgical approach and predicting a patient’s long-term prognosis.
Treatment:
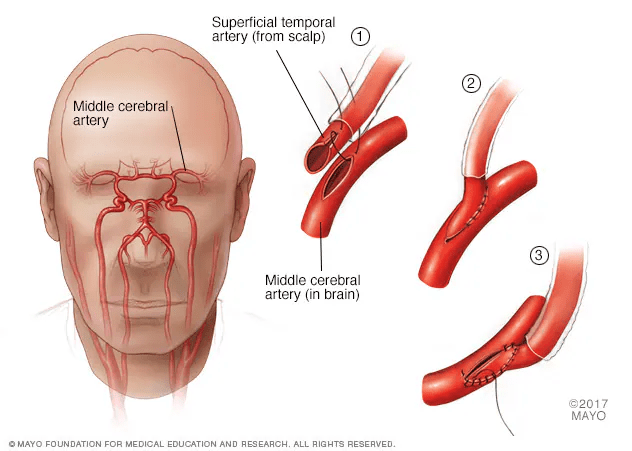
Moyamoya disease is a progressive condition that cannot be halted by medication alone, making surgical revascularization the definitive treatment for symptomatic patients. While aspirin and cilostazol are frequently used to manage blood clot risks and improve post-surgical vessel health, they do not address the underlying arterial narrowing. Conversely, certain treatments like long-term anticoagulants and specific migraine medications are generally avoided due to the risks of brain hemorrhage or further reduced blood flow. To minimize stroke risk, patients must focus on maintaining stable blood pressure, proper hydration, and avoiding anemia.
The primary surgical objective is to restore blood supply by redirecting scalp vessels to the brain through direct, indirect, or combined bypass techniques. Clinical evidence, such as the Japan Adult Moyamoya Trial, highlights the efficacy of these procedures in reducing recurrent bleeding and ischemic strokes. The long-term outcomes are particularly striking in pediatric cases, where surgery can reduce the risk of stroke from 90% down to less than 5%. Because endovascular options like stenting carry high risks and low success rates, expert consensus emphasizes treatment at high-volume specialized centers and genetic counseling for families with hereditary patterns.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop Moyamoya disease. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about Moyamoya disease, donation opportunities, or the progress being made on potential treatments, visit the MoyaMoya Foundation.
Let’s keep spreading awareness! – Lily
References:
Smith, E. R., & Scott, R. M. (2023, February 9). Moyamoya Disease – Symptoms, Causes, & Treatment | NORD. NORD (National Organization for Rare Disorders). https://rarediseases.org/rare-diseases/moyamoya-disease/

Leave a comment