What is Acquired Aplastic Anemia?:
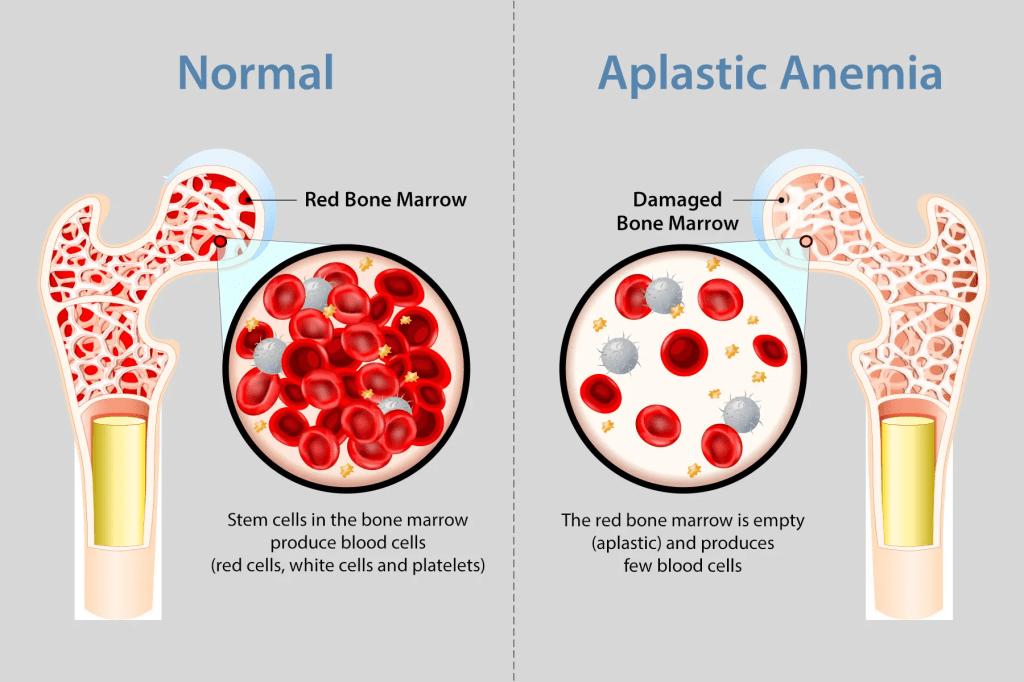
Acquired aplastic anemia is a rare and serious blood disorder characterized by bone marrow failure, where the spongy tissue inside large bones, like those of the spine, pelvis, and legs, fails to produce adequate blood cells. Normally, the hematopoietic stem cells within the bone marrow undergo self-renewal and differentiate into red blood cells, white blood cells, and platelets. In acquired aplastic anemia, an almost complete absence of these stem cells leads to severely low levels of all three blood cell types, resulting in anemia, bleeding, and even infection. While bone marrow failure can sometimes be secondary to other disorders or stem from inherited genetic mutations that first manifest in adulthood without a family history, most cases are autoimmune in nature, occurring when the immune system mistakenly targets the bone marrow. Consequently, the standard treatment for most patients involves immunosuppressive therapies, typically utilizing antithymocyte globulin and cyclosporine. This disease is classified as severe based on specific blood counts, and while patients with moderate decreases may not require immediate treatment, severe cases require intervention.
Symptoms:
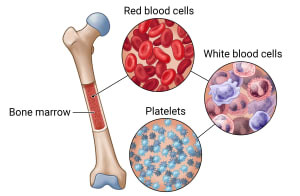
The symptoms of acquired aplastic anemia stem directly from the bone marrow’s inability to produce adequate blood cells, leading to a trio of deficiencies known as anemia, leukopenia, and thrombocytopenia. Anemia triggers fatigue, weakness, dizziness, pale skin, shortness of breath, and potentially chest pain. Meanwhile, a lack of infection-fighting white blood cells leaves individuals highly susceptible to bacterial and fungal infections, and a shortage of clot-forming platelets results in easy bruising, frequent nose or gum bleeds, and abnormally heavy menstrual cycles. The overall severity and progression of these symptoms vary wildly, ranging from mild, stable conditions to sudden, life-threatening complications.
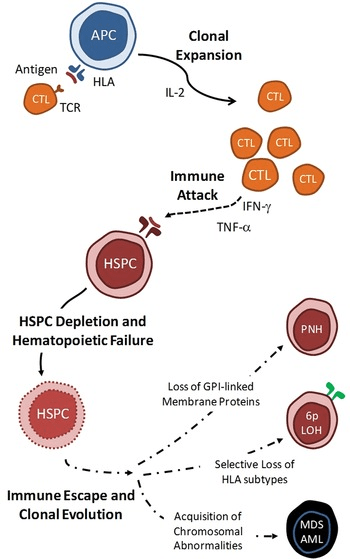
Furthermore, acquired aplastic anemia shares a complex, close relationship with other bone marrow disorders, most notably paroxysmal nocturnal hemoglobinuria (PNH). PNH is caused by an acquired genetic mutation in the PIGA gene of marrow stem cells, making blood cells vulnerable to destruction by the body’s own complement immune proteins; roughly half of aplastic anemia patients show evidence of PNH at diagnosis or during recovery following immunosuppressive therapy. Additionally, individuals face a risk of their condition evolving into myelodysplasia (MDS), and, in a minority of cases, the disease may ultimately progress into leukemia.
Causes:
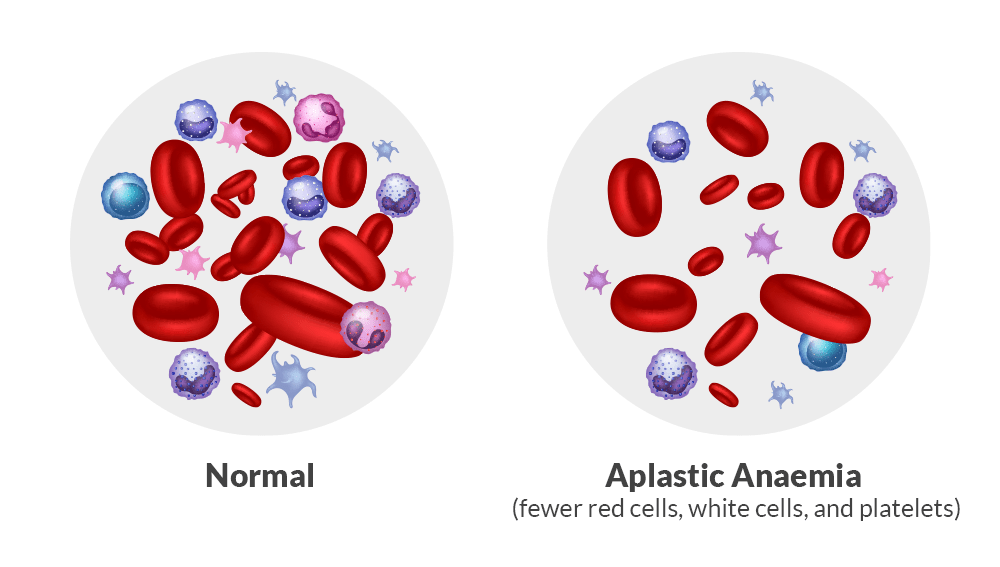
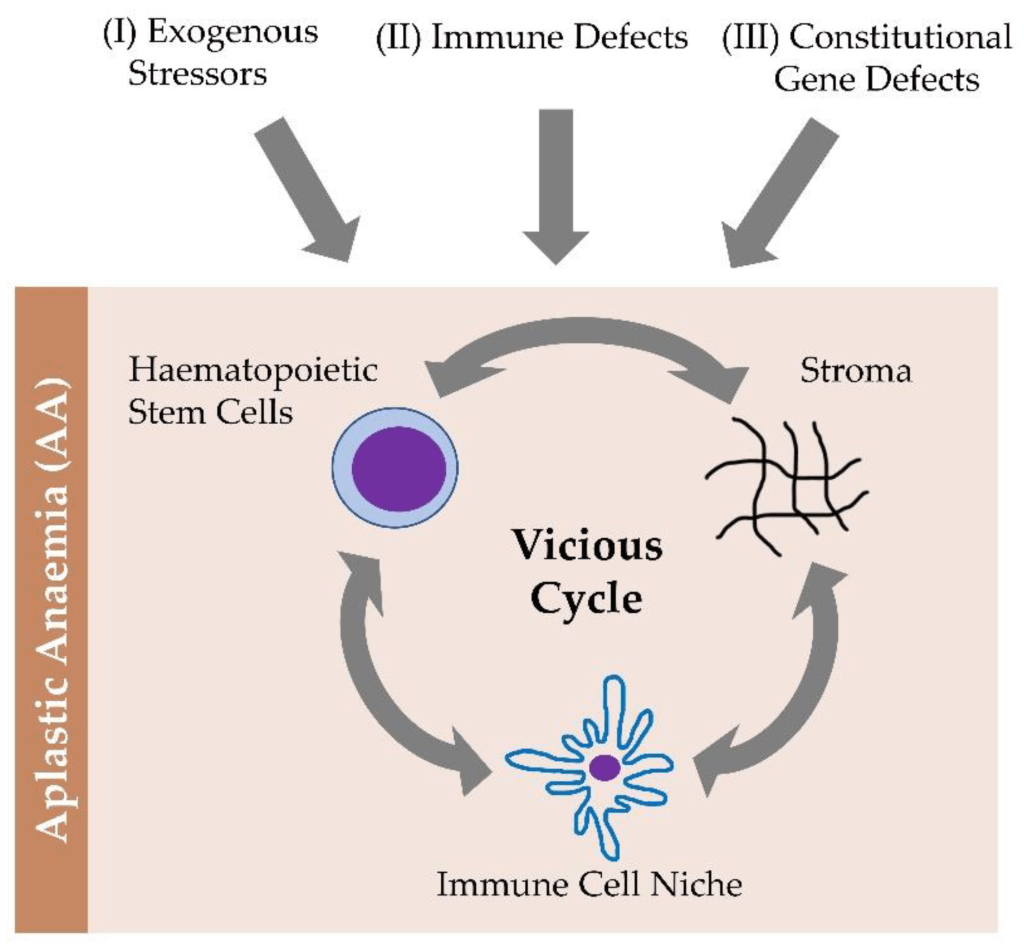
Acquired aplastic anemia occurs mostly for unknown reasons, although strong evidence suggests it is driven by an autoimmune response. In this process, the body’s immune system, specifically T-lymphocytes, mistakenly targets and destroys hematopoietic stem cells in the bone marrow. Consequently, the bone marrow is depleted of these primitive cells, often appearing replaced by fat, which ultimately leads to a severe deficiency of all three blood cell types, a condition known as pancytopenia.
While the exact trigger for this immune-mediated destruction has remained unidentifiable, the disease has been linked to certain environmental factors. Historically, exposure to benzene has been connected to direct bone marrow damage, while other chemicals like pesticides and nonviral hepatitis are rarely associated but hypothesized to occasionally spark the destructive immune response. Despite these known potential triggers, the vast majority of acquired aplastic anemia cases develop without any identifiable environmental cause.
Diagnosis:
A diagnosis of acquired aplastic anemia is typically suspected when an otherwise healthy individual presents with pancytopenia, which is a marked deficiency in all three types of blood cells. To confirm the diagnosis, physicians conduct a thorough clinical evaluation and detailed patient history alongside specialized testing, most notably a bone marrow biopsy. During this procedure, a small tissue sample is surgically extracted from the hip or pelvis and examined under a microscope, where it can reveal a dramatic reduction or complete absence of blood-producing cells. Additional testing is usually required to rule out alternative underlying conditions, such as leukemia, and to verify that the bone marrow failure does not stem from an inherited or genetic cause.
Treatment:
Treatment for acquired aplastic anemia is tailored to a patient’s age, overall health, and disease severity, focusing on both immediate symptom relief and correcting the underlying bone marrow failure. Initial management relies on supportive care, including transfusions and antibiotics. For a definitive cure, allogeneic bone marrow transplantation, where a patient’s diseased marrow is destroyed via chemotherapy and replaced with healthy marrow from a matched relative or unrelated donor, is the preferred treatment for children and younger adults. While highly effective, transplants carry risks of graft rejection and graft-versus-host disease (GVHD), which can range from mild to life-threatening and require management with specialized medications.
For older patients or those who lack a suitable donor, immunosuppressive therapy serves as the primary treatment strategy. By utilizing drugs like cyclosporine and antithymocyte globulin (ATG), this approach suppresses the immune system, allowing the bone marrow to recover and resume blood cell production. Although immunosuppression can restore blood counts for extended periods, the improvement is not always permanent, requiring repeated treatment during relapses.
For the approximately one-third of patients who do not respond to initial immunosuppression, stem cell transplantation or repeated courses of immunosuppressive drugs may be considered. While traditional hematopoietic growth factors are ineffective, the platelet-production stimulator eltrombopag has proven highly successful for refractory cases. Eltrombopag is approved for patients with severe aplastic anemia who fail immunosuppression and cannot undergo a transplant, and combining it with standard immunosuppressive drugs as a first-line therapy has been shown to significantly boost both overall and complete response rates.
How You Can Make an Impact:
Without proper research, funding, and support for continued studies and clinical trials to determine possible cures, legitimate medicines for the disease, or preventative treatment, many more people will go on to develop acquired aplastic anemia. If you can, please donate here! If you are unable to donate, consider volunteering your time by raising awareness for this rare disease. If you’re interested in learning more about acquired aplastic anemia disease, donation opportunities, or the progress being made on potential treatments, visit the Aplastic Anemia & MDS International Foundation.
Let’s keep spreading awareness! – Lily
References:
Young, N. S. (2018, June 20). Acquired Aplastic Anemia – Symptoms, Causes, Treatment | NORD. NORD (National Organization for Rare Disorders); NORD. https://rarediseases.org/rare-diseases/acquired-aplastic-anemia/

Leave a comment